
Abstract
Background and aims
Central obesity is a condition that poses a significant risk to global health and requires the employment of novel scientific methods for exploration. The objective of this study is to use DNA methylation analysis to detect DNA methylation loci linked to obesity phenotypes, i.e. waist circumference and waist-to-hip ratio adjusted for BMI.
Methods and results
Two-hundred and ten healthy European participants from the STANISLAS Family Study (SFS), comprising 73 nuclear families, were comprehensively assessed for methylation status using Illumina Infinium HumanMethylation450 BeadChip. An epigenome-wide association study was performed, which identified a CpG site cg16170243 located on chromosome 18q21.2 significantly associated with waist circumference, after adjusting for BMI (β = 2.32, SE = 0.41, Padj = 0.048). Cg16170243 corresponds to a 50 bp-length human methylation oligoprobe located within the AC090241.2 gene that overlaps ST8SIA5 gene. No significant association was observed with waist-to-hip ratio adjusted for BMI (Padj > 0.05).
Conclusions
A novel association between DNA methylation and WC was identified, which is demonstrating that epigenetic mechanisms may have a significant impact on waist circumference ratio in healthy individuals. Further studies are warranted to address the causal effects of this association.
Similar content being viewed by others
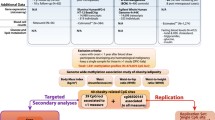

Background
Epigenetic changes are covalent modifications of cytosine bases, histones and changes in nucleosome positioning that can modulate the effect of a genotype on a particular phenotype and thus affect physiological mechanisms as well as the pathophysiology of many diseases [1]. The most widely studied among them is cytosine methylation [2], a covalent attachment of methyl group to a DNA sequence that generally results in silencing of genes encoded in the methylated region [3]. DNA methylation patterns are not static but undergo precise, highly coordinated changes that can be mediated both by environmental and genetic factors and inherited through mitotic cell divisions [4]. This process occurs already during embryogenesis and is crucial for development, differentiation and cellular variability [3], as well as for the transcriptional regulation of genes and miRNA [5]. On the other hand, atypical patterns of DNA methylation are associated with obesity, oxidative stress, hypertension, inflammation, angiogenesis and other pathological processes that are implicated in the development of chronic diseases [6].
Obesity is a systemic disease and a growing health problem that contributes to the increased risk of many common medical conditions [7]. The excess of adipose tissue provokes chronic low-grade inflammation, associated with immunological activation and oxidative stress, insulin resistance, hypertension and dyslipidemia [8]. The most common approach to determine general obesity is body mass index (BMI), the ratio between the mass (weight) and height of an individual, expressed in kg/m2. However, clinical evidence suggests that diseases, such as diabetes, are more associated with central obesity, where visceral adipose tissue is stored [9]. Therefore, waist circumference (WC) and waist-to-hip ratio can well account for obesity-related pathologies [10].
Increasing prevalence of obesity worldwide is mostly due to changes in the environment, whereas a person’s genetic profile is considered one of the main causes of individual difference in predisposition to weight gain. A high heritability of this phenotype has been confirmed in several epidemiological studies [11] and examined in genome-wide association studies (GWAS), where 27–30% of the total BMI variance was explained by common single nucleotide polymorphisms (SNPs) [12].
Furthermore, obesogenic environment during in uterus development and in early childhood was associated with an increased risk of a range of chronic diseases in adulthood, showing that early environmental influences can cause permanent effects [13]. This interaction of biological and environmental factors is believed to be mediated by epigenetic mechanisms, by which the environmental factors could change gene expression and thus explain the increased prevalence of obesity in the last few decades [12,13,14]. Epigenome-wide association studies (EWAS) gave the possibility of in-depth insight into epigenetic changes [15]. Several EWAS were performed to study DNA methylation to identify the common variation in the DNA methylome, related to obesity phenotypes, which pointed out several loci, i.e. HIF3A [8] ABCG1 and CPT1A [8, 16].
In order to make further contribution to the comprehension of obesity trait, in this study, we are examining the methylation levels associated with central obesity, measured by WC and waist-to-hip ratio adjusted for BMI, in a healthy population. Using healthy individuals can help to avoid the discrepancies caused by extremes of obesity and comorbidity in population-specific cohorts. Our findings provide new insights into genetic regulation of visceral fat accumulation and are presenting a new variant, which may increase the susceptibility to chronic diseases.
Methods
Population
210 healthy related individuals from 73 families of the STANISLAS Family Study (SFS) have been enrolled in this study, including 115 adults and 95 children. The SFS is a 10-year longitudinal cohort that includes three visits at 5-year intervals. It involved 1,006 French families from Vandoeuvre-lès-Nancy, France, first that were first recruited between 1993–1995 [17]. All subjects were of European-Caucasian origin, without the presence of chronic disorders (CVD, cancer, diabetes, hypertension etc.). Descriptive characteristics are presented in Table 1.
The study protocols were approved by the institutional ethics committees and all subjects gave written informed consent for their participation in the study. For the participants of less than 18 years old, consent was also given for their participation by their parents. Since SFS is a cohort of families, the parents and their children participated in the study and after being informed by the researchers, they all gave written consent for their participation and the participation of their children.
Data collection
Data and blood samples were collected during the baseline visit of the SFS. All measurements of the clinical indicators were performed in the laboratory of the Centre for Preventive Medicine (CMP) in Vandoeuvre lès Nancy, France. Blood count was performed using standard methodology in the laboratory of the CMP. WC was taken at the midpoint between the lower margin of the last palpable rib and the top of the iliac crest (hip bone). Trained professionals recorded measurements to the nearest 0.1 cm. BMI was calculated by the Quetelet index formula as weight (kg) divided by height2 (m)2. Data collection has been previously described [17, 18]. Obesity was defined as BMI ≥ 30 kg/m2 for adults and for children BMI ≥ 97th percentile for age and sex calculated on French BMI curves [19].
DNA methylation assay and quality control
Blood samples were taken in the morning between 8 and 9 a.m. following an overnight fast. Whole blood DNA was extracted by the Miller technique [20] and was stored at − 80 °C. Genome-wide DNA methylation profiling was performed using the Infinium HumanMethylation450 BeadChip (Illumina). Methylation ratio (referred to as beta value by Ilumina’s software), is the proportion of methylated by all CpGs (methylated / (methylated + unmethylated) CpGs). Methylation arrays were analyzed and visualized using the R package minfi (version 1.16.1) [21]. Detection p-value was generated for every CpG in all samples, indicating the quality of the signal. Poor quality probes were excluded from the analysis using a detection p-value cutoff (˃0.05). Probes, missing in more than 5% of samples were excluded from all samples. Background correction and normalization were performed with Illumina background correction and SWAN [22] to all intensity values for a total of 485 512 probes.
We further excluded probes already annotated in HumanMethlyation450 annotation files (probes containing SNPs, sex chromosomes, and a single base extension (SBE) sites). Finally, the probes containing cross-reactive and target polymorphic CpGs were excluded. All downstream analyses were carried out with software R.
Genotyping and selection of SNPs
Genotyping was performed by the Infinium CoreExome Illumina assay. Significant WC-associated SNPs, located in the same chromosome as the significant methylation sites were selected from the NHGRI-EBI GWAS catalog [23]. These SNPs were extracted from the GWAS data and used in association analyses as candidate genes.
Statistical analysis
Waist-to-hip ratio and WC were not normally distributed and were therefore transformed to the e-log scale. Individual analyses have been performed for the adult population, the children population and the combined population (children and adults together). A linear mixed-effects model was used to analyze the association between methylation levels at each probe and log-transformed phenotypes. The model used included sex, age, BMI, family structure, and individual white cell counts (neutrophils, lymphocytes, monocytes, eosinophils and basophils) as covariates and chip array as random effect. Bonferroni correction and false discovery rate (FDR) methods (< 0.05) were used for the correction of the results for multiple testing. The association analyses of methylation values and the assessed phenotypes were performed by using the package CpGassoc in R [24]. HumanMethlyation450 annotation file was used for annotating the probes and their corresponding genes. Deviation of multiple correlation squared ρ2 from constant (random model) of G*Power software was used in order to calculate statistical power [23], specifically the post hoc power analysis procedure. This procedure is parameter analysis which requires the type of test (tails: one or two), effect size (H1 ρ2 and H0 ρ2), α error probability, total sample size, and number of predictors.
In order to investigate whether the observed association between WC and methylation level was related to genetic variants associated with WC, a linear mixed-effects model with age, sex, BMI, family structure, methylation batch array and white cell counts as fixed effects, and methylation chip as a random effect was carried out for association analysis using the R statistical package nlme [25].
In silico analysis
The significant CpGs were localized on the Human genome (GRCh38.p12) using Ensembl browser. In silico gene expression was obtained using the BLUEPRINT Data Analysis portal [26] and GTEx Portal [27].
Results
Poor quality probes (n = 764) were excluded from the sample. In total, 77% of probes passed quality control, excluding probes containing SNPs, sex chromosomes, SBE sites, cross-reactive and target polymorphic CpGs, thus leaving 373 626 probes for association analyses (Fig. 1). The post hoc analysis of the statistical power of the result was calculated as 100%.

Quality control (QC) processing
One individual was excluded after quality control checks of the methylation array data (outlier of the plotted median of the methylated against unmethylated samples), thus 210 participants were included in the analyses.
The results of the analysis showed one novel significant positive association of cg16170243 probe with WC adjusted for sex, age, BMI, family structure, and individual blood cell counts (β = 2.32, SE = 0.41; Padj = 0.048) in the combined population. QQ plot of the EWAS is presented in Fig. 2. CpG site cg16170243 (chr18:46759502-46759551) corresponds to a 50 bp-length human methylation oligoprobe located on chromosome 18q21.1 (Table 2). No significant associations were identified for waist-to-hip ratio.

Quantile–quantile plot (QQ plot) of the distribution of observed log10 association p-values against the expected null distribution, lambda = 0.937
Three SNPs, previously associated with WC in GWAS [28], were identified on chromosome 18 (rs6567160, rs7239883, rs12970134). Minor allele frequencies (MAF) were 0.23, 0.38 and 0.25 accordingly and were in agreement with the MAF in 1000G. No significant associations were identified between these polymorphisms and methylation of the cg16170243.
The cg16170243 probe maps on chromosome 18q21.1 and is located in the forward strand of the ST8SIA5 antisense gene (AC090241.2), whereas in the opposite strand the probe is located 245 base pairs upstream of the ST8SIA5 gene (Fig. 3). The ST8SIA5 antisense gene has 3 splice variants: AC090241.2-201, AC090241.2-202 and AC090241.2-203. Cg16170243 is located both within an intron of AC090241.2-202 and 173 base pairs upstream of AC090241.2-203 (Fig. 3). In silico analysis showed that cg16170243 is embedded in the promoter flank of the ST8SIA5 gene (ENSR00000575029) and contains transcription factor binding sites (TFBS).

Environment of cg16170243 probe. As depicted by Fig. 3, cg16170243 (in green) is located on chromosome 18q21.1 within intron of AC090241.2-202 transcript and 173 base pairs upstream of the AC090241.2-203 transcript of the forward strand, and 245 base pairs upstream of ST8SIA5 gene in the opposite strand. Furthermore, cg16170243 is embedded in the promoter flank of ST8SIA5 gene (ENSR00000575029, in red) and overlaps 8 TFBS (in grey)
Analysis of the expression data showed that ST8SIA5 gene is expressed in several cell-types of the bloodline: neutrophils, peripheral blood mononuclear cells, eosinophils and monocytes (Fig. 4). Furthermore, the additional data confirmed the expression of ST8SIA5 in blood cells and indicated that the gene is to a greater extent expressed also in brain structures (Fig. 5).

In silico expression of ST8SIA5 gene and AC090311.1 pseudogene

In silico expression of ST8SIA5 gene in brain and whole blood
Discussion
We have identified a novel significant positive association (β = 2.32, SE = 0.41; Padj = 0.048) between WC, adjusted for BMI, and methylation at the probe cg16170243. There was no association of cg16170243 with three candidate SNPs previously associated with WC. Therefore, the underlying mechanism of the CpG’s effect on WC is probably differentiated from the mechanisms linked to these genetic variants.
The ST8SIA5 antisense gene has 3 splice variants. Probe cg16170243 is located both in the intronic zone of AC090241.2-202 variant and at the 5’ of AC090241.2-203 variant (Fig. 3). AC090241.2-202 splice variant is an antisense biotype transcript, meaning that it overlaps the genomic span of a protein-coding locus on the opposite strand and can be an important regulator of gene expression [29]. The overlapping gene on the opposite strand of AC090241.2-202 is ST8SIA5 (Alpha-2,8-sialyltransferase 8E) gene, coding for a sialyltransferase enzyme, involved in the synthesis of gangliosides GD1c, GT1a and GQ1b from GM1b, GD1a and GT1b, respectively [30]. Gangliosides are glycosphingolipids with one or more sialic acids. They are a component of the bilayer lipid membrane on the cell surface, where they present the points of recognition for extracellular molecules on surfaces of neighboring cells and serve for interaction between cells, adhesion, cell differentiation and transduction of signal [31]. Cg16170243 methylation may modify expression levels of ST8SIA5 which would lead to an affected synthesis of gangliosides.
Studies showed that an inadequate ganglioside expression in mediobasal hypothalamic neurons deregulates neuronal leptin [32] and insulin signaling [33], which can affect body weight regulation and energy homeostasis. Gangliosides interact with molecules of signal transduction pathways, such as receptors tyrosine kinases (RTKs). Change in ganglioside composition induces the dissociation of RTKs from glycolipid-enriched microdomains, which results in reduced phosphorylation of the receptors and thus causes insulin resistance [34]. Accordingly, insulin is a critical regulator of adipocyte biology and resistance of insulin receptors is, on the one hand, one of the important causes of obesity, and on the other hand, one of the biggest contributors to the development of obesity [35].
Therefore, it is plausible that the effect of cg16170243 on ST8SIA5 gene in blood cells reflects a process that also occurs on a larger scale in neuronal cells when methylation at cg16170243 site is present, causing a disruption of the insulin signaling pathway and contributing to the accumulation of visceral fat (Fig. 5). Nevertheless, further studies that would confirm such hypothesis are required.
Despite the above-proposed mechanisms of the methylation impact on the WC via insulin deregulation, we cannot exclude the possibility that methylation could be a consequence of modified WC, rather than a cause. Indeed, a meta-analysis from S. Wahl et al. has shown that variation in DNA methylation is most often a consequence of adiposity [36]. Thus, particular molecular mechanisms could enable the methylation of regulatory regions of genes involved in obesity, but the exact mechanisms of this regulation remain to be elucidated.
Epigenetics studies of obese phenotypes on healthy individuals have been previously done [37], however, this is in our knowledge the only EWAS study including only healthy subjects. The post hoc power analysis has shown that the statistical power of our result was very high (100%). It should be mentioned, however, that analysis on separate children and adult populations was also performed and no significant associations were identified, probably due to decreased power of the smaller samples of the separate analyses. Similarly, we have performed separate analysis by sex and again no significant associations were identified.
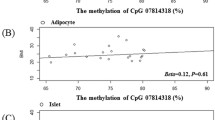
A limitation of our study is the small sample size. Although we have identified a significant result with high statistical power, we did not have the possibility to replicate it in a population-specific study. As epigenetic changes can be tissue-specific, the limitation of our study is also the use of blood samples without tissue-specific replications. Even though the within-subject correlation of CpG specific sites from blood and adipose tissue was previously confirmed, the use of methylation markers in blood to mirror the corresponding profile in the target tissue should be taken with caution [38].
Conclusions
We identified a novel association between DNA methylation and WC. This association could be due to the modification of the regulatory region of ST8SIA5 transcription, resulting in a perturbed synthesis of gangliosides.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the Open Science Framework (OSF) repository at https://mfr.osf.io/render?url=https%3A%2F%2Fosf.io%2F4h5v7%2Fdownload.
Abbreviations
- BMI:
-
Body mass index
- CVD:
-
Cardiovascular disease
- DNA:
-
Deoxyribonucleic acid
- EWAS:
-
Epigenome-wide association studies
- FDR:
-
False discovery rate
- GWAS:
-
Genome-wide association studies
- PBMC:
-
Peripheral blood mononuclear cell
- RTKs:
-
Receptors tyrosine kinases
- SBE:
-
Single base extension
- SD:
-
Standard deviation
- SE:
-
Standard error
- SFS:
-
STANISLAS Family Study
- SNP:
-
Single nucleotide polymorphism
- TFBS:
-
Transcription factor binding sites
- WC:
-
Waist circumference
References
Dupont C, Armant DR, Brenner CA. Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med. 2009;27(5):351–7.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68.
Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13(7):484–92.
Feil R, Fraga MF. Epigenetics and the environment: emerging patterns and implications. Nat Rev Genet. 2012;13(2):97–109.
Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet. 2007;16 Spec No 1:R50-9.
Levy E, Spahis S, Bigras JL, Delvin E, Borys JM. The epigenetic machinery in vascular dysfunction and hypertension. Curr Hypertens Rep. 2017;19(6):52.
Abdelaal M, le Roux CW, Docherty NG. Morbidity and mortality associated with obesity. Ann Transl Med. 2017;5(7):161.
Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou YH, et al. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015;24(15):4464–79.
Vazquez G, Duval S, Jacobs DR Jr, Silventoinen K. Comparison of body mass index, waist circumference, and waist/hip ratio in predicting incident diabetes: a meta-analysis. Epidemiol Rev. 2007;29:115–28.
WHO. Waist Circumference and Waist–Hip Ratio: Report of a WHO Expert Consultation 2008.
Albuquerque D, Stice E, Rodriguez-Lopez R, Manco L, Nobrega C. Current review of genetics of human obesity: from molecular mechanisms to an evolutionary perspective. Mol Genet Genom MGG. 2015;290(4):1191–221.
Pigeyre M, Yazdi FT, Kaur Y, Meyre D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin Sci. 2016;130(12):943–86.
Bell CG. The epigenomic analysis of human obesity. Obesity (Silver Spring, Md). 2017;25(9):1471–81.
Cordero P, Li J, Oben JA. Epigenetics of obesity: beyond the genome sequence. Curr Opin Clin Nutr Metab Care. 2015;18(4):361–6.
Flanagan JM. Epigenome-wide association studies (EWAS): past, present, and future. Methods Mol Biol (Clifton, NJ). 2015;1238:51–63.
Aslibekyan S, Demerath EW, Mendelson M, Zhi D, Guan W, Liang L, et al. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring, Md). 2015;23(7):1493–501.
Visvikis-Siest S, Siest G. The STANISLAS Cohort: a 10-year follow-up of supposed healthy families. Gene–environment interactions, reference values and evaluation of biomarkers in prevention of cardiovascular diseases. Clin Chem Lab Med. 2008;46(6):733–47.
Siest G, Visvikis S, Herbeth B, Gueguen R, Vincent-Viry M, Sass C, et al. Objectives, design and recruitment of a familial and longitudinal cohort for studying gene-environment interactions in the field of cardiovascular risk: the Stanislas cohort. Clin Chem Lab Med. 1998;36(1):35–42.
Rolland-Cachera MF, Cole TJ, Sempé M, Tichet J, Rossignol C, Charraud A. Body Mass Index variations: centiles from birth to 87 years. Eur J Clin Nutr. 1991;45(1):13–21.
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, et al. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics (Oxford, England). 2014;30(10):1363–9.
Maksimovic J, Gordon L, Oshlack A. SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 2012;13(6):R44.
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019;47(D1):D1005–12.
Barfield RT, Kilaru V, Smith AK, Conneely KN. CpGassoc: an R function for analysis of DNA methylation microarray data. Bioinformatics (Oxford, England). 2012;28(9):1280–1.
Pinheiro J, Bates D, DebRoy S, Sarkar D, R Core Team. nlme: linear and nonlinear mixed effects models. 2017.
Fernandez JM, de la Torre V, Richardson D, Royo R, Puiggros M, Moncunill V, et al. The BLUEPRINT data analysis portal. Cell Syst. 2016;3(5):491-5.e5.
Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (New York, NY). 2015;348(6235):648–60.
MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017;45(D1):D896-d901.
Pelechano V, Steinmetz LM. Gene regulation by antisense transcription. Nat Rev Genet. 2013;14(12):880–93.
Kim YJ, Kim KS, Do S, Kim CH, Kim SK, Lee YC. Molecular cloning and expression of human alpha2,8-sialyltransferase (hST8Sia V). Biochem Biophys Res Commun. 1997;235(2):327–30.
Yu RK, Tsai YT, Ariga T, Yanagisawa M. Structures, biosynthesis, and functions of gangliosides—an overview. J Oleo Sci. 2011;60(10):537–44.
Nordstrom V, Willershauser M, Herzer S, Rozman J, von Bohlen Und Halbach O, Meldner S, et al. Neuronal expression of glucosylceramide synthase in central nervous system regulates body weight and energy homeostasis. PLoS Biol 2013;11(3):e1001506.
Herzer S, Meldner S, Grone HJ, Nordstrom V. Fasting-induced lipolysis and hypothalamic insulin signaling are regulated by neuronal glucosylceramide synthase. Diabetes. 2015;64(10):3363–76.
Julien S, Bobowski M, Steenackers A, Le Bourhis X, Delannoy P. How do gangliosides regulate RTKs signaling? Cells. 2013;2(4):751–67.
Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Investig. 2000;106(4):473–81.
Wahl S, Drong A, Lehne B, Loh M, Scott WR, Kunze S, et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature. 2016;541:81.
Boyne DJ, Friedenreich CM, McIntyre JB, Courneya KS, King WD. Associations between adiposity and repetitive element DNA methylation in healthy postmenopausal women. Epigenomics. 2017;9(10):1267–77.
Huang Y-T, Chu S, Loucks EB, Lin C-L, Eaton CB, Buka SL, et al. Epigenome-wide profiling of DNA methylation in paired samples of adipose tissue and blood. Epigenetics. 2016;11(3):227–36.
Acknowledgements
None.
Funding
Financial support was provided by the European Union within the frames of the Operational Programme FEDER-FSE Lorraine et Massif des Vosges 2014–2020, by Agence Nationale de la Recherche, programme d’Investissements d’avenir, grant number ANR-15RHU-0004. Additionally, this work was supported by the regional project CPER-ITM2P 2015–2020.
Author information
Authors and Affiliations
Contributions
TX, VG and MGS participated in study design, performed data analysis and interpretation and drafted the manuscript. SD participated in bioinformatics analysis and drafting of the manuscript. EWAS analysis pipeline was designed by EM and PD. CM prepared biological material and experiments. HM, JL and PF contributed with biological measurements. SVS was involved in the project conception, development of overall research plan, and study oversight, and provided the necessary administrative, technical and material support for the research. All authors participated in the interpretation of data and review of the manuscript. They all approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Study protocols were approved by the institutional ethics committee CCPPRB de Lorraine (Comité consultatif de protection des personnes dans la recherche biomédicale) and CNIL (Commission Nationale de l'Informatique et des Libertés). All subjects gave written informed consent for their participation in the study. For participants < 18 years old written consent has been provided from them and their parents who also participated in the study and were thus fully informed about the aims and the research protocols.
Consent for publication
There are no identifying images or other personal or clinical details of participants presented that compromise anonymity. All data are anonymous. All participants have given their written consent for participating in the study and publish anonymous results. For participants < 18 years old written consent has been provided from them and their parents who also participated in the study and were thus fully informed about the aims and the research protocols.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Peer review reports.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xie, T., Gorenjak, V., Stathopoulou, M.G. et al. Epigenome-wide association study detects a novel loci associated with central obesity in healthy subjects. BMC Med Genomics 14, 233 (2021). https://doi.org/10.1186/s12920-021-01077-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01077-9