
Abstract
Background
Chordoma is a rare bone tumor that is typically resistant to chemotherapy and is associated with genetic abnormalities of the T-box transcription factor T (TBXT) gene, which encodes the transcription factor brachyury. Brachyury is felt to be a major contributor to the development of chordomas.
Case presentation
We describe a 67-year-old woman who developed an undifferentiated pleomorphic sarcoma in her thigh. Despite treatment with standard chemotherapy regimens, she had a rapidly progressive course of disease with pulmonary metastases and passed away 8 months from diagnosis with pulmonary complications. Her medical history was remarkable in that she had a spheno-occipital chordoma at age 39 and later developed multiple other tumors throughout her life including Hodgkin lymphoma and squamous cell carcinoma and basal cell carcinoma of the skin. She had a family history of chordoma and her family underwent extensive genetic study in the past and were found to have a duplication of the TBXT gene.
Conclusions
Brachyury has been found to associate with tumor progression, treatment resistance, and metastasis in various epithelial cancers, and it might play roles in tumorigenesis and aggressiveness in this patient with multiple rare tumors and germ line duplication of the TBXT gene. Targeting this molecule may be useful for some malignancies.
Similar content being viewed by others

Background
Chordoma is a rare bone sarcoma with an incidence rate below 0.1 per 100,000 [1]. It is derived from remnants of the notochord, an embryonic structure that is required for the induction of the neural plate in the embryonic disk. Chordoma typically occurs in the skull base, mobile spine, and sacrum. Although distant metastasis may occur, chordomas usually behave as low-grade neoplasms with a locally aggressive growth pattern and high local recurrence rates. Surgery and radiotherapy are the mainstays of treatment, but many patients develop tumor recurrence or complications from treatment. These tumors typically are resistant to traditional chemotherapy and no standard treatment has been approved [2, 3].
While most cases of chordoma are sporadic, reports of two or more close relatives with chordoma suggest a genetic predisposition for this disease. Probable autosomal dominant inheritance in familial chordoma was first reported by Stepanek et al. [4]. A series of subsequent studies demonstrated duplication of the TBXT gene, a member of the T-box proteins encoding brachyury, that is felt to be a major susceptibility mechanism for the development of chordoma in several families [5, 6]. Brachyury is a transcription factor within the T-box family of genes that is expressed in the nuclei of notochord cells and is essential for notochord development [7]. Knocking down brachyury in a chordoma cell line resulted in decreased proliferation and cell senescence [8]. Brachyury is considered a marker for notochord and notochord-derived tumors with nearly a 100% expression rate, although it has been reported to be expressed in some germ cell tumors and small cell lung cancer [9, 10].
In this report, we described a patient with a history of familial chordoma who later developed multiple cancers including squamous cell carcinoma (SCC) of the skin, basal cell carcinoma (BCC) of the skin, Hodgkin lymphoma, and aggressive undifferentiated pleomorphic sarcoma (UPS). Genetic mechanisms underlying the pathogenesis of familial chordoma and multiple cancers are discussed.
Case presentation
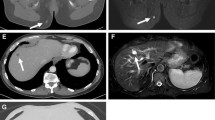
A 67-year-old white woman developed gradually increasing right hip and thigh pain over three months. Physical examination revealed a 20 × 10 cm medial and posterior soft tissue mass in right thigh with moderate tenderness to palpation. Motor power and sensation were intact. An MRI showed an 18.6 × 13 × 11-cm mass within the hamstring musculature (Fig. 1). A biopsy revealed a high-grade UPS (Fig. 2), and a PET-CT revealed bilateral hypermetabolic lung nodules and right inguinal chain hypermetabolic lymphadenopathy consistent with metastatic disease. She was treated with pegylated liposomal doxorubicin (PLD) and infusional ifosfamide with mesna [11]. Repeat imaging demonstrated resolution of the lung nodules after 1 cycle. However, after the 3rd cycle, imaging revealed progression in the primary tumor (21 × 20 × 30-cm) (Fig. 3) and multiple new pulmonary metastases. The primary tumor was surgically excised for symptom control; pathology of the resection specimen revealed a FNCLCC grade 3 UPS with lymphovascular invasion and negative margins with a chemotherapy effect in ~ 60% of the tumor (Fig. 4).

Pre-treatment MRI images. A Axial PD fat suppressed MR image of the right thigh demonstrates a heterogeneous soft tissue mass measuring 13.5 × 11.4 cm in transverse and AP dimension; B Coronal stir (short T1 inversion recovery) MR image of a heterogenous soft tissue mass measuring 20.8 × 11.3 cm in craniocaudal and transverse measurement

Core needle biopsy specimen showing tumor heterogeneity with high cellularity on the left and stromal collagenization on the right (A). Higher magnification image demonstrates a high-grade pleomorphic sarcoma with severe nuclear atypia (B). Microscopy was performed using an Olympus BX46 microscope with UPlanFL N lenses and an Olympus DP73 camera with no filter; acquisition software was Olympus cellSens standard with a resolution of 4800 (W) × 3600(H) pixels and no downstream processing

Post-treatment MRI images. Axial PD fat suppressed MR image of the right thigh demonstrates interval increase in the size of the soft tissue mass, measuring 21.6 × 20.9 cm in transverse and AP dimension

The sarcoma shows very high cellularity with a sheet-like growth pattern, multinucleated giant cells and necrosis (A). On higher magnification, there are high-grade pleomorphic tumor cells and multiple mitotic figures (B) with areas of heterologous osteosarcomatous differentiation (C, right). Chemotherapy effect, characterized by cell death and hyalinization, was present in ~ 60% of the tumor (D, right). Microscopy was performed as in Fig. 2
Next generation sequencing (NGS) using the FoundationOne platform of the primary tumor revealed several genetic changes including missense mutation of AXL (R368Q) and RB1 (R661W) and intron 5 rearrangement of FAS. The tumor was microsatellite stable and had a tumor mutational burden of 3 mutations/Mb. In addition, several variants of unknown significance were identified, including T535N in ALK, R496H in BRCA1, L2277F in BRCA2, Q740H in BRIP1, S301F in CCT6B, L219I in CSF3R, S1134C in CUX1, R127Q in ETV6, P197L in IL7R, L168* in JAK3, K2148N in MKI67, I754M in MSH3, G1366S in NOTCH1, and ROS1 rearrangement. The TBXT gene, which encodes brachyury, is not included in the FoundationOne testing (Additional file 1: Technical specifications of FoundationOne NGS platform. Genes examined and mutations detected). Immunostaining of the primary UPS specimen revealed no detectable nuclear brachyury staining (Fig. 5). Due to disease progression and performance status, treatment with gemcitabine was initiated [12]. The patient only received 1 cycle of treatment before she developed acute hypoxic respiratory failure with bilateral pleural effusions and left pneumothorax, with clinical and imaging findings of disease progression. The treatment regimen was changed to pembrolizumab plus pazopanib [13,14,15]; although well-tolerated, she developed continued tumor progression and passed away 8 months after diagnosis.

Immunohistochemistry stain for Brachyury is negative in the tumor cells (A); a positive control shows brachyury expression in a chordoma (B). Microscopy was performed as in Fig. 2
Her past medical history was notable for a spheno-occipital chordoma excised at age 39. Interestingly, ten members of her family were diagnosed with chordoma. Of ten cases, nine involved the clivus or nasopharynx with the age of diagnosis ranging from 6 to 68-years-old. One brother was diagnosed at age 28 with an aggressive sacral chordoma and later died of metastatic disease. The patient and members of her family were involved in extensive genetic studies to identify the genetic abnormality in familial chordoma, which showed TBXT gene duplication on 6q27 and contained a SNP variant rs2305089 [5, 6]. Her last follow-up MRI, at age 61, still revealed a stable 1.2 × 0. 8 × 2.3-cm soft tissue mass localized in the posterior nasopharynx appearing to connect a midline defect in the clivus. There was no evidence of progression or metastatic disease, and clinical observation was recommended. There was no history of second malignancy in her other family members with chordoma.
At age 52 she was diagnosed with SCC, which was incidentally found in the pathology specimen after hemorrhoidectomy. This was an invasive carcinoma treated with further surgical excision with no adjuvant therapy. Follow-up high-resolution anoscopy with biopsies revealed no evidence of any residual dysplasia or carcinoma in situ. She also was diagnosed with several superficial BCC lesions, which were removed during the previous 15 years before her death. She also had an adenomatous colonic polyp removed at age 59. At age 62 she developed a gradually enlarging right neck mass over 2 months. A biopsy revealed classical Hodgkin disease, nodular sclerosing type, and PET-CT imaging showed bilateral hypermetabolic supraclavicular, mediastinal, and left hilar adenopathy (stage IIA). She was treated with four cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine as the ABVD regimen; PET-imaging after two cycles revealed a complete response. She completed chemotherapy followed by radiation therapy to the mediastinum, left hilar, and supraclavicular areas. She had no subsequent evidence of lymphoma recurrence. She also had a benign PET-negative thyroid nodule found during work-up for lymphoma that remained stable. She had no significant exposure history, worked in an office, and was a never smoker.
Discussion and conclusions
We describe a patient with a familial chordoma and a history of multiple cancers throughout her life including Hodgkin disease and UPS. The UPS progressed rapidly on PLD/ifosfamide and subsequent treatments with gemcitabine and then pembrolizumab plus pazopanib. We discuss the biology of chordoma and the use of genetic studies to broaden treatment options for aggressive tumors that do not respond to standard therapy.
Genomic rearrangements, including copy number variants, contribute to disease susceptibility in both sporadic as well as some inherited Mendelian diseases. The patient and members of her family were involved in genetic studies to identify the genetic abnormality underlying the pathogenesis of familial chordoma. High-resolution array-CGH (comparative genomic hybridization) revealed that her rare variant of chordoma is associated with TBXT gene duplication on 6q27 and TBXT gene sequencing showed an SNP variant rs2305089 (G177D). TBXT encodes brachyury, which is important in notochord development and expressed in most sporadic chordomas. All affected individuals in the family shared a common 6q disease-related haplotype. [5, 6]. These findings are in line with data from other studies that previously identified brachyury as a crucial factor in the pathogenesis of chordoma [16,17,18]. Furthermore, brachyury has been reported to be a driver of cancer stemness and therapy resistance, and may play roles in cancer progression, epithelial-mesenchymal transition (EMT), and metastasis in various types of cancer [19,20,21] including breast cancer [22,23,24,25], prostate cancer [26, 27], non-small cell lung cancer [28,29,30], colorectal cancer [31], hepatocellular carcinoma [32], and some other epithelial cancers [33,34,35]. There is no known association between T gene and the other tumors (SCC, BCC, Hodgkin lymphoma, or UPS) that occurred in the patient described here. The mechanism was proposed to be through the Yes-associated protein (YAP) regulatory axis, a key regulator of tissue growth and homeostasis [36]. Amplification of the YAP gene locus has been reported in a wide spectrum of human and murine tumors, and one study found that brachyury enhances YAP transcription by binding with the proximal promoter region to increase its stability post-transcription [37].
UPS is one of the most common subtypes of soft tissue sarcoma. It is characterized by a lack of definite lineage differentiation using currently available diagnostic techniques, and studies suggest several subtypes of soft tissue sarcoma, including liposarcoma and leiomyosarcoma, may evolve into UPS [38]. UPS is a disease with complex genomic alterations, and gene expression studies suggest the existence of functional subgroups of UPS that have different metastatic propensity and clinical outcomes [39,40,41]. The most commonly identified mutated genes in UPS were TP53 (66%), ATRX genes (34%), and RB1 (28%); although the frequency varies from study to study, there are consistent trends among these 3 genes [42,43,44]. Nonetheless, there are very few clinically targetable mutations identified to date [45].
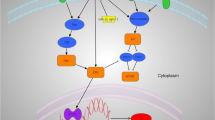
Given her known status of TBXT gene duplication, brachyury might play a role in both the development and the aggressiveness and resistance to therapy of the UPS in her case (Fig. 6). Therapy that has demonstrated efficacy in pre-clinical studies of chordoma include afatinib, an epidermal growth factor receptor (EGFR) inhibitor, and THZ1, a cyclin-dependent kinase (CDK) 7/12/13 inhibitor [46, 47]. Afatinib was the only EGFR inhibitor that inhibited the proliferation of all chordoma cell lines tested, and its antiproliferative activity correlated with the ability to promote degradation of EGFR and brachyury [46]. CDK inhibitors targeting CDK7/12/13 and CDK9 have also been found to suppress chordoma cell proliferation, reduce tumor growth in vivo, and decrease brachyury protein expression in these systems [47]. The transcription-associated CDKs, including CDK7, CDK8, CDK9, CDK12 and CDK13, are important regulators of gene expression [48], and transcription-associated CDK inhibitors have been found to down-regulate highly expressed, enhancer-associated transcription factors in other cancers [47]. TBXT is associated with a 1.5 Mb region containing strong enhancers or “super-enhancers,” and is the most highly expressed super-enhancer associated transcription factor in chordomas [47]. Therefore, transcription-associated CDK inhibitors may exert their action by down-regulating brachyury [47, 49], providing an example of transcription factor down-regulation by a small molecule. Various strategies to target brachyury are currently under investigation in clinical trials (Table 1). Results of early phase trials of a brachyury vaccine demonstrated induction of an immune responses to brachyury and showed some evidence of clinical benefit in patients with chordoma and metastatic solid tumors [50,51,52].

Potential effects of brachyury in promoting cancer aggressiveness. Over-expression of brachyury promotes cancer stemness and resistance to chemotherapy and radiation, and also enhances epithelial-mesenchymal transition, migration, invasion, and, eventually, metastasis. The mechanism is not well defined, but may relate to up-regulation of YAP transcription and stabilization of YAP protein by increased production of brachyury, with subsequent effects mediated primarily by YAP. This is an original figure
In a panel of soft tissue sarcomas, 0/60 UPS cases not associated with chordoma were found to have nuclear expression of brachyury. Interestingly, in one study, 75/76 chordomas had nuclear brachyury expression, while the one negative case exhibited sarcomatous transformation. Thus, the finding that our UPS case did not express brachyury does not exclude that it could have arisen from a chordoma, although the location of the tumor would be very atypical [10]. On a tissue-based NGS study (FoundationOne®), the UPS tumor of the patient described here demonstrated microsatellite stability, low tumor mutational burden, and mutations in AXL and RB1. AXL activation could predict resistance to EGFR inhibitors [55, 56]. However, no known clinical significance on the effect of the AXL missense mutation in this case is known. RB1 inactivation, predicted by a missense mutation in the pocket domain (aa 773–928) as seen in this case, may be associated with sensitivity of Aurora kinase A and resistance to CDK4/6 inhibitors, but this is also not clinically targetable at present [57]. ROS1 rearrangement, also found in our patient’s tumor, is a common event in carcinogenesis and has been demonstrated in a variety of human cancers, including glioblastoma, non-small cell lung cancer (NSCLC), and sarcomas, such as angiosarcoma and epithelioid hemangioendothelioma [58]. Thus, her tumor may have been responsive to ROS1 tyrosine kinase inhibitors, such as crizotinib, although she did not receive that trial agent; most of the studies to date have focused on lung cancer models [59, 60]. Interestingly, despite being the most common gene with genetic alteration in UPS, there was no TP53 mutation found on the FoundationOne platform in this patient (Additional file 1). Several variants of unknown significance were also detected as described above, but their association with malignancy is currently unknown.
In this patient with multiple rare tumors including a UPS with an aggressive nature, multiple genetic alterations such as AXL and RB1 mutation might play a role. However, the occurrence of multiple uncommon tumors suggests an underlying susceptibility, and the presence of the germline TBXT duplication may have an important role in the pathogenesis of her tumors and their biology. Detailed molecular and genetic studies could offer therapeutic targets to alleviate the progression of disease in the future.
Availability of data and materials
Not applicable as all data is shown in Additional file 1.
Abbreviations
- BCC:
-
Basal cell carcinoma
- CDK:
-
Cyclin-dependent kinase
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- FNCLCC:
-
French Federation of Cancer Centers Sarcoma Group
- MRI:
-
Magnetic resonance imaging
- NGS:
-
Next-generation sequencing
- NSCLC:
-
Non-small cell lung cancer
- PET-CT:
-
Positron emission tomography-computed tomography
- PLD:
-
Pegylated liposomal doxorubicin
- SCC:
-
Squamous cell carcinoma
- SNP:
-
Single-nucleotide polymorphism
- TBXT:
-
T-box transcription factor T
- UPS:
-
Undifferentiated pleomorphic sarcoma
- YAP:
-
Yes-associated protein
References
McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12(1):1–11.
Smoll NR, Gautschi OP, Radovanovic I, Schaller K, Weber DC. Incidence and relative survival of chordomas: the standardized mortality ratio and the impact of chordomas on a population. Cancer. 2013;119(11):2029–37.
Alan O, Akin Telli T, Ercelep O, Tanrikulu Simsek E, Basoglu Tuylu T, Mutis A, et al. Chordoma: a case series and review of the literature. J Med Case Reports. 2018;12(1):239.
Stepanek J, Cataldo SA, Ebersold MJ, Lindor NM, Jenkins RB, Unni K, et al. Familial chordoma with probable autosomal dominant inheritance. Am J Med Genet. 1998;75(3):335–6.
Yang XR, Ng D, Alcorta DA, Liebsch NJ, Sheridan E, Li S, et al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet. 2009;41(11):1176–8.
Kelley MJ, Shi J, Ballew B, Hyland PL, Li WQ, Rotunno M, et al. Characterization of T gene sequence variants and germline duplications in familial and sporadic chordoma. Hum Genet. 2014;133(10):1289–97.
Kispert A, Ortner H, Cooke J, Herrmann BG. The chick Brachyury gene: developmental expression pattern and response to axial induction by localized activin. Dev Biol. 1995;168(2):406–15.
Presneau N, Shalaby A, Ye H, Pillay N, Halai D, Idowu B, et al. Role of the transcription factor T (brachyury) in the pathogenesis of sporadic chordoma: a genetic and functional-based study. J Pathol. 2011;223(3):327–35.
Vujovic S, Henderson S, Presneau N, Odell E, Jacques TS, Tirabosco R, et al. Brachyury, a crucial regulator of notochordal development, is a novel biomarker for chordomas. J Pathol. 2006;209(2):157–65.
Miettinen M, Wang Z, Lasota J, Heery C, Schlom J, Palena C. Nuclear Brachyury expression is consistent in chordoma, common in germ cell tumors and small cell carcinomas, and rare in other carcinomas and sarcomas: an immunohistochemical study of 5229 cases. Am J Surg Pathol. 2015;39(10):1305–12.
Skubitz KM, Wilson JD, Cheng EY, Lindgren BR, Boylan KLM, Skubitz APN. Effect of chemotherapy on cancer stem cells and tumor-associated macrophages in a prospective study of preoperative chemotherapy in soft tissue sarcoma. J Transl Med. 2019;17(1):130.
Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, Samuels B, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol. 2007;25(19):2755–63.
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2012;379(9829):1879–86.
Tawbi HA, Burgess M, Bolejack V, Van Tine BA, Schuetze SM, Hu J, et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): a multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017;18(11):1493–501.
Monga V, Skubitz KM, Maliske S, Mott SL, Dietz H, Hirbe AC, et al. A retrospective analysis of the efficacy of immunotherapy in metastatic soft-tissue sarcomas. Cancers (Basel). 2020;12(7):1873.
Hu Y, Mintz A, Shah SR, Quinones-Hinojosa A, Hsu W. The FGFR/MEK/ERK/brachyury pathway is critical for chordoma cell growth and survival. Carcinogenesis. 2014;35(7):1491–9.
Zhu J, Kwan KM, Mackem S. Putative oncogene Brachyury (T) is essential to specify cell fate but dispensable for notochord progenitor proliferation and EMT. Proc Natl Acad Sci USA. 2016;113(14):3820–5.
Yan X, Li Z, Li H, Liu P, Zhao Z, Cheng S, et al. Inhibition of glycogen synthase kinase 3 beta suppresses the growth and survival of Skull base chordoma cells by downregulating brachyury expression. Onco Targets Ther. 2019;12:9783–91.
Fernando RI, Litzinger M, Trono P, Hamilton DH, Schlom J, Palena C. The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells. J Clin Invest. 2010;120(2):533–44.
Hamilton DH, Huang B, Fernando RI, Tsang KY, Palena C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer Res. 2014;74(9):2510–9.
Hamilton DH, McCampbell KK, Palena C. Loss of the cyclin-dependent kinase inhibitor 1 in the context of brachyury-mediated phenotypic plasticity drives tumor resistance to immune attack. Front Oncol. 2018;8:143.
Shao C, Zhang J, Fu J, Ling F. The potential role of Brachyury in inducing epithelial-to-mesenchymal transition (EMT) and HIF-1α expression in breast cancer cells. Biochem Biophys Res Commun. 2015;467(4):1083–9.
Li K, Ying M, Feng D, Du J, Chen S, Dan B, et al. Brachyury promotes tamoxifen resistance in breast cancer by targeting SIRT1. Biomed Pharmacother. 2016;84:28–33.
Li K, Ying M, Feng D, Chen Y, Wang J, Wang Y. SMC1 promotes epithelial-mesenchymal transition in triple-negative breast cancer through upregulating Brachyury. Oncol Rep. 2016;35(4):2405–12.
Chen M, Zou S, He C, Zhou J, Li S, Shen M, et al. Transactivation of SOX5 by Brachyury promotes breast cancer bone metastasis. Carcinogenesis. 2020;41:551–60.
Pinto F, Pértega-Gomes N, Pereira MS, Vizcaíno JR, Monteiro P, Henrique RM, et al. T-box transcription factor brachyury is associated with prostate cancer progression and aggressiveness. Clin Cancer Res. 2014;20(18):4949–61.
Pinto F, Pértega-Gomes N, Vizcaíno JR, Andrade RP, Cárcano FM, Reis RM. Brachyury as a potential modulator of androgen receptor activity and a key player in therapy resistance in prostate cancer. Oncotarget. 2016;7(20):28891–902.
Huang B, Cohen JR, Fernando RI, Hamilton DH, Litzinger MT, Hodge JW, et al. The embryonic transcription factor Brachyury blocks cell cycle progression and mediates tumor resistance to conventional antitumor therapies. Cell Death Dis. 2013;4(6):e682.
Xu K, Liu B, Liu Y. Impact of Brachyury on epithelial-mesenchymal transitions and chemosensitivity in non-small cell lung cancer. Mol Med Rep. 2015;12(1):995–1001.
Wan Z, Jiang D, Chen S, Jiao J, Ji L, Shah AS, et al. T-box transcription factor brachyury promotes tumor cell invasion and metastasis in non-small cell lung cancer via upregulation of matrix metalloproteinase 12. Oncol Rep. 2016;36(1):306–14.
Jezkova J, Williams JS, Jones-Hutchins F, Sammut SJ, Gollins S, Cree I, et al. Brachyury regulates proliferation of cancer cells via a p27Kip1-dependent pathway. Oncotarget. 2014;5(11):3813–22.
Du R, Wu S, Lv X, Fang H, Wu S, Kang J. Overexpression of brachyury contributes to tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma. J Exp Clin Cancer Res. 2014;33(1):105.
Shimoda M, Sugiura T, Imajyo I, Ishii K, Chigita S, Seki K, et al. The T-box transcription factor Brachyury regulates epithelial-mesenchymal transition in association with cancer stem-like cells in adenoid cystic carcinoma cells. BMC Cancer. 2012;12:377.
Kobayashi Y, Sugiura T, Imajyo I, Shimoda M, Ishii K, Akimoto N, et al. Knockdown of the T-box transcription factor Brachyury increases sensitivity of adenoid cystic carcinoma cells to chemotherapy and radiation in vitro: implications for a new therapeutic principle. Int J Oncol. 2014;44(4):1107–17.
Akimoto N, Nakamura K, Hijioka H, Kume K, Matsumura Y, Sugiura T. Transfection of T-Box transcription factor BRACHYURY and SOX2 synergistically promote self-renewal and invasive phenotype in oral cancer cells. Int J Mol Sci. 2018;19(11):3620.
Shah SR, David JM, Tippens ND, Mohyeldin A, Martinez-Gutierrez JC, Ganaha S, et al. Brachyury-YAP regulatory axis drives stemness and growth in cancer. Cell Rep. 2017;21(2):495–507.
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19(4):491–505.
Skubitz KM, Skubitz AP. Characterization of sarcomas by means of gene expression. J Lab Clin Med. 2004;144(2):78–91.
Chibon F, Lagarde P, Salas S, Pérot G, Brouste V, Tirode F, et al. Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med. 2010;16(7):781–7.
Skubitz KM, Francis P, Skubitz AP, Luo X, Nilbert M. Gene expression identifies heterogeneity of metastatic propensity in high-grade soft tissue sarcomas. Cancer. 2012;118(17):4235–43.
Skubitz KM, Skubitz AP, Xu WW, Luo X, Lagarde P, Coindre JM, et al. Gene expression identifies heterogeneity of metastatic behavior among high-grade non-translocation associated soft tissue sarcomas. J Transl Med. 2014;12:176.
Pérot G, Chibon F, Montero A, Lagarde P, de Thé H, Terrier P, et al. Constant p53 pathway inactivation in a large series of soft tissue sarcomas with complex genetics. Am J Pathol. 2010;177(4):2080–90.
Cancer Genome Atlas Research Network. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell. 2017;171(4):950-965.e28.
Zheng B, Qu Y, Wang J, Shi Y, Yan W. Pathogenic and targetable genetic alterations in resected recurrent undifferentiated pleomorphic sarcomas identified by targeted next-generation sequencing. Cancer Genomics Proteomics. 2019;16(3):221–8.
Lewin J, Garg S, Lau BY, Dickson BC, Traub F, Gokgoz N, et al. Identifying actionable variants using next generation sequencing in patients with a historical diagnosis of undifferentiated pleomorphic sarcoma. Int J Cancer. 2018;142(1):57–65.
Magnaghi P, Salom B, Cozzi L, Amboldi N, Ballinari D, Tamborini E, et al. Afatinib is a new therapeutic approach in chordoma with a unique ability to target EGFR and Brachyury. Mol Cancer Ther. 2018;17(3):603–13.
Sharifnia T, Wawer MJ, Chen T, Huang QY, Weir BA, Sizemore A, et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat Med. 2019;25(2):292–300.
Chou J, Quigley DA, Robinson TM, Feng FY, Ashworth A. Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 2020;10(3):351–70.
Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511(7511):616–20.
Heery CR, Singh BH, Rauckhorst M, Marté JL, Donahue RN, Grenga I, et al. Phase I trial of a yeast-based therapeutic cancer vaccine (GI-6301) targeting the transcription factor Brachyury. Cancer Immunol Res. 2015;3(11):1248–56.
Heery CR, Palena C, McMahon S, Donahue RN, Lepone LM, Grenga I, et al. Phase I study of a poxviral TRICOM-based vaccine directed against the transcription factor Brachyury. Clin Cancer Res. 2017;23(22):6833–45.
Collins JM, Donahue RN, Tsai YT, Manu M, Palena C, Gatti-Mays ME, et al. Phase I trial of a modified vaccinia Ankara priming vaccine followed by a Fowlpox virus boosting vaccine modified to express Brachyury and costimulatory molecules in advanced solid tumors. Oncologist. 2020;25(7):560-e1006.
Hu Y, Singh R, Deng Z, Mintz A, Hsu W. Liposome-protamine-DNA nanoparticle-mediated delivery of short hairpin RNA targeting brachyury inhibits chordoma cell growth. J Biomed Nanotechnol. 2016;12(10):1952–61.
Cottone L, Cribbs AP, Khandelwal G, Wells G, Ligammari L, Philpott M, et al. Inhibition of histone H3K27 demethylases inactivates Brachyury (TBXT) and promotes chordoma cell death. Cancer Res. 2020;80(20):4540–51.
Dufies M, Jacquel A, Belhacene N, Robert G, Cluzeau T, Luciano F, et al. Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget. 2011;2(11):874–85.
Zhang Z, Lee JC, Lin L, Olivas V, Au V, LaFramboise T, et al. Activation of the AXL kinase causes resistance to EGFR-targeted therapy in lung cancer. Nat Genet. 2012;44(8):852–60.
Oser MG, Fonseca R, Chakraborty AA, Brough R, Spektor A, Jennings RB, et al. Cells Lacking the RB1 tumor suppressor gene are hyperdependent on aurora B kinase for survival. Cancer Discov. 2019;9(2):230–47.
Davies KD, Doebele RC. Molecular pathways: ROS1 fusion proteins in cancer. Clin Cancer Res. 2013;19(15):4040–5.
Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371(21):1963–71.
Mazières J, Zalcman G, Crinò L, Biondani P, Barlesi F, Filleron T, et al. Crizotinib therapy for advanced lung adenocarcinoma and a ROS1 rearrangement: results from the EUROS1 cohort. J Clin Oncol. 2015;33(9):992–9.
Acknowledgements
We thank the James Dinerstein and Kevin Franklin families for research support, Michael Franklin for editorial assistance, and Wannasiri Chiraphapphaiboon, MS for illustrating Fig. 6.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
NS created an outline, wrote the manuscript, and designed Fig. 6. PM reviewed and interpreted pathology slide and provided consultation on pathology results, SM reviewed and interpreted MRI and PET-CT images and provided consultation on imaging results, DRC provided consultation on the clinical, imaging, and surgical perspective of the disease, KMS devised the project and its outline, and supervised the project overall. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable; the University of Minnesota allows single case reports without formal review.
Consent for publication
Written consent obtained from the patient.
Competing interests
The authors declare that they have no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Technical specifications of FoundationOne NGS platform. Genes examined and mutation detected.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sumransub, N., Murugan, P., Marette, S. et al. Multiple malignant tumors in a patient with familial chordoma, a case report. BMC Med Genomics 14, 213 (2021). https://doi.org/10.1186/s12920-021-01064-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-01064-0