
Abstract
Background
Kidney renal clear cell carcinoma (KIRC) is the most common type of kidney cell carcinoma which has the worst overall survival rate. Almost 30% of patients with localized cancers eventually develop to metastases despite of early surgical treatment carried out. MicroRNAs (miRNAs) play a critical role in human cancer initiation, progression, and prognosis. The aim of our study was to identify potential prognosis biomarkers to predict overall survival of KIRC.
Methods
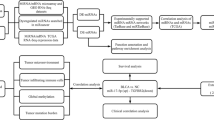
All data were downloaded from an open access database The Cancer Genome Atlas. DESeq2 package in R was used to screening the differential expression miRNAs (DEMs) and genes (DEGs). RegParallel and Survival packages in R was used to analysis their relationships with the KIRC patients. David version 6.8 and STRING version 11 were used to take the Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis.
Results
We found 2 DEGs (TIMP3 and HMGCS1) and 3 DEMs (hsa-miR-21-5p, hsa-miR-223-3p, and hsa-miR-365a-3p) could be prognosis biomarkers for the prediction of KIRC patients. The constructed prognostic model based on those 2 DEGs could effectively predict the survival status of KIRC. And the constructed prognostic model based on those 3 DEMs could effectively predict the survival status of KIRC in 3-year and 5-year.
Conclusion
The current study provided novel insights into the miRNA related mRNA network in KIRC and those 2 DEGs biomarkers and 3 DEMs biomarkers may be independent prognostic signatures in predicting the survival of KIRC patients.
Similar content being viewed by others


Background
Kidney Cancer is one of the most common malignancies which account for 2.2% of all new cancer cases and 1.8% of all cancer related death in 2018 globally [1]. Renal cell carcinoma (RCC) accounts for 90% of all kidney cancers [2]. RCC could be divided into three categories: kidney renal clear cell carcinoma (KIRC), kidney renal papillary cell carcinoma (KIRP) and kidney chromophobe (KICG). Among them, KIRC is the most common type which accounts for about 70–75% of RCC [2]. Due to the radiotherapy and chemotherapy resistance, surgical treatment is currently the most effective way for KIRC patients [3]. However previous studies indicated that KIRC usually has the worst overall survival rate [2]. Almost 30% of patients with localized cancers eventually develop to metastases despite of early surgical treatment carried out [4]. Therefore, it is necessary to identify suitable prognosis biomarkers for the diagnosis and treatment of KIRC even though many prognosis factors have been reported for KIRC.
Cancer stem cells are a subpopulation of cells that has the driving force of carcinogenesis which is a complex and multistep phenomenon and involves accumulation of genetic and epigenetic changes ultimately leading to the development of pathological manifestations [5]. MicroRNAs (miRNAs) are a family of endogenous, small, noncodingRNAs. MiRNAs could regulate the expression of genes associated with various biological phenomenons such as homeostasis, development, proliferation, differentiation, and apoptosis [6, 7]. Deregulated expression and signaling of miRNA have been well-studied in the pathogenesis of various cancers. Aberrant expressions of miRNAs are vital for the initiation and progression of human malignancies as they act as both tumor suppressors and oncogenes [8]. In the present study, we aimed to identify potential prognosis biomarkers to predict overall survival of KIRC.
Methods
Data source and data processing
KIRC (71 controls vs 516 cancers) miRNA sequencing data, KIRC (72 controls vs 530 cancers) mRNA sequencing data and the corresponding clinical information were obtained from an open database TCGA. DESeq2 was used to identify the DEMs and DEGs according to |log2FC| > 0.5, basemean > 50, padj < 0.05. And we utilized two different web based tools, miRDB and TargetScanHuman 7.2, to screen the target genes of miRNAs. The target genes only enriched in two databases could be selected as putative target genes for the next analysis.
Gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) pathway enrichment analysis
DAVID version 6.8 was used to determine the association among target genes. To gain insight into the biological functions of those DEGs, GO and KEGG pathway enrichment analyses were performed.
Protein–protein interaction (PPI) network and module analysis
STRING version 11 was used to assess PPI information. A normal medium confidence interval of 0.4 was used as threshold. Cytoscape_v3.7.2 software was used to visualize the resulting PPI network. The Molecular Complex Detection (MCODE) application was used to select significant modules from the PPI network in Cytoscape_v3.7.2.
Pathologic TNM correlation analysis
After the classification base on the size and/or extent of the main cancer (T1 + 2 and T3 + 4), lymph node metastasis (N0 and N1), and distant metastasis (M0 and M1) respectively, we used Cox regression analysis, Kaplan–Meier curve, and log-rank analysis to further verify the characteristics of the pathologic TNM and the intensity of their correlation with survival. A repeated-measure ANOVA followed by Bonferroni post hoc tests or unpaired two-tail Student’s t test was used exam the correlation of DEGs with pathologic TNM.
Survival analysis
Median is the number in the middle of a set of data in order. We divided the samples into high expression group and low expression group based on the median. We used RegParallel and survival packages in R to carry out univariate and multivariate Cox regression analysis.
Specific prognostic model construction
After multivariate Cox regression analysis, we constructed specific prognostic models according to previous reports [9]. KIRC patients were divided into low risk group and high risk group depend on the median value of the risk score. Patients whose risk values were higher than the median were classified as high risk, and Patients whose risk values were lower than the median were classified as low-risk. And then we used survival analysis to know the relationship of the models with the survival of KIRC patient. And then we constructed time-dependent receiver operating characteristic (ROC) curves within 1-, 3-, and 5-year and estimated its utility as a prognostic model for predicting the survival status.
Results
Identification of DEMs and DEGs
TCGA is an open access database containing miRNA/mRNA profiles and the corresponding clinical information. Using Padj < 0.05 and |log2FC|> 0.5 as cut-off criteria for DEMs, 111 DEMs were identified by DESeq2 analysis, including 62 up-regulated DEMs and 59 down-regulated DEMs (Fig. 1a). Using basemean > 50, Padj < 0.05 and |log2FC| > 0.5 as cut-off criteria for DEGs, 8694 DEGs were identified by DESeq2 analysis, including 5288 up-regulated DEMs and 3406 down-regulated DEMs (Fig. 1b). Then we performed survival analysis for those 111 DEMs and found that 40 DEMs were correlated with the overall survival of KIRC patients. Of which 25 DEMs was up-regulated and 15 DEMs was down-regulated (Table 1).

Differential miRNAs and mRNAs expression analysis. a Volcano plot of DEMs for KIRC. b Volcano plot of DEGs for KIRC. c Scatter plot of Log2FC(miRNA) versus Log2FC(mRNA) in KIRC. d The number of miRNAs-mRNAs verified by correlation analysis
To ensure the integrity of the target genes, we utilized miRDB and TargetScanHuman7.2 for target gene prediction. Following integrated analysis of DEGs and target genes of DEMs, a total of 1874 pairs miRNAs-mRNAs were identified involved 1364 DEGs, including 647 up-regulated DEGs and 717 down-regulated DGEs (Fig. 1c). Since the relationship between DEGs and DEMs is negatively correlated, we introduced the spearman correlation analysis by using p < 0.05 and r < − 0.3 as cut-off criteria. And we obtained 267 pairs miRNAs-mRNAs which contained 20 DEMs (such as, Top 3 up-regulated DEMs: hsa-miR-155-5p, hsa-miR-21-5p, and hsa-miR-584-5p. Top 3 down-regulated DEMs: hsa-miR-204-5p, hsa-miR-30c-2-3p, and hsa-miR-30a-3p) and 252 DEGs (such as, Top 3 up-regulated DEMs: IGLON5, SOX11, and LOX. Top 3 down-regulated DEMs: EHF, MUC15, and CALB1) (Fig. 1d).
GO function and KEGG pathway enrichment analysis of the DEGs.
To gain a deeper understanding of the selected DEGs, we performed GO and KEGG analysis for those 252 DEGs. There were 65 biological process (BP), 23 cellular component (CC), and 29 molecular functions (MF) that were enriched by GO analysis (such as, Top 3 GO-BP, signal transduction, negative regulation of transcription from RNA polymerase II promoter, and positive regulation of transcription from RNA polymerase II promoter. Top 3 GO-CC, plasma membrane, endoplasmic reticulum, and cell surface. Top 3 GO-MF, protein binding, ATP binding, and transcription factor activity, sequence-specific DNA binding) (Fig. 2a–c, Additional file 1: Table S1). And there were 20 KEGG pathways that were enriched by KEGG analysis, of which 9 signaling pathway was enriched significantly (such as Top 3 signaling pathway, hsa04144: Endocytosis, hsa04510: Focal adhesion, and hsa04914: Progesterone-mediated oocyte maturation) (Fig. 2d, Additional file 1: Table S2).

Functional enrichment analysis and PPI network construction. a–c The significantly enriched top 10 GO term (p value < 0.05) analyzed by David 6.8 for KIRC. d The significantly enriched KEGG pathway (p value < 0.05) analyzed by David 6.8 for KIRC. e PPI network of DEGs with their degree higher than the average in KIRC. f PPI network of DEGs verified by MCODE analysis. The orange represents un-regulated genes; the green represents down-regulated genes
PPI network construction and module selection.
Using the STRING database and Cytoscape software, a total of 252 DEGs were filtered into the PPI network, containing 252 nodes and 274 edges. According to the view that highly connected genes can have a major impact on disease, we identified 86 DEGs (with degree > average 2.17) as high connectivity genes in our study (Fig. 2e). According to their degree of importance, 10 important modules involved 43 DEGs from the PPI network complex were selected for further analysis based on Cytoscape MCODE (Fig. 2f). By cross analysis of those 86 DEGs and those 43 DEGs, we identified 10 overlap DEGs (ANXA1, BCL11B, CYCS, HLA-G, HMGCS1, RAB5C, RNF123, TIMP3, TRPM4, ZEB2).
Pathologic TNM correlation analysis
We classified the pathologic TNM staging of KIRC patients and conducted an overall survival correlation analysis. The results indicated that pathologic TNM were actually correlated with the overall survival. The KIRC patients with bigger of the size and/or extent of the main cancer (T3 + T4), or lymph node metastasis (N1), or distant metastasis (M1) displayed the worse overall survival rate (Fig. 3a–c). Subsequently, we evaluated their relationship of those 10 overlap DEGs with pathologic TNM, and found that 5 DEGs was confirmed to be associated with pathologic T and pathologic M (Fig. 3d–e).

Pathologic TNM correlation analyses. a–c Survival curves of pathologic TNM for KIRC. d Associated analyses of DEGs with pathologic T stage for KIRC. e Associated analyses of DEGs with pathologic M stage for KIRC. *p < 0.05, **p < 0.01, ***p < 0.001
Specific prognostic model construction
After pathologic TNM correlation analysis, we performed univariate Cox regression analysis for those 10 DEGs. We found that HMGCS1, TIMP3, and RNF123 were correlated with overall survival. The KIRC patients with high expression of HMGCS1, TIMP3, and RNF123 exhibited a better overall survival (Fig. 4a–c). Then, we performed multivariate Cox regression analysis for HMGCS1, TIMP3, and RNF123. And the result showed that TIMP3 (p = 0.0001) and HMGCS1 (p = 0.0110) were still correlated with the overall survival of KIRC patients, of which the expression of TIMP3 (logFC = − 0.766, p < 0.001) and HMGCS1 (logFC = − 0.643, p < 0.001) were decreased significantly.

Construction of survival risk score system based on DEGs signature. a–c Survival curves of DEGs in KIRC. d Survival curves of prognostic model based on DEGs in KIRC. e–g The ROC curve of prognostic model based on DEGs in 1-, 3-, and 5-year with AUC value
Followed the multivariate Cox regression analysis, we constructed a prognostic model by using TIMP3 and HMGCS1. The patients with low risk actually exhibited a better overall survival (Fig. 4d). The time-dependent receiver operating characteristic (ROC) curves had area under curve (AUC) values higher than 0.5, which were 0.7487, 0.6893, and 0.6893 respectively (Fig. 4e–g).
We screen the negative related miRNAs of TIMP3 and HMGCS1 among those 20 DEMs, and found TIMP3 was correlated with hsa-miR-21-5p (r = − 0.520), HMGCS1 were correlated with hsa-miR-223-3p (r = − 0.318) and hsa-miR-365a-3p (r = − 0.340). The result of multivariate Cox regression analysis showed that hsa-miR-21-5p [p = 0.0002, HR = 1.854(1.343–2.560)], hsa-miR-223-3p [p = 0.0002, HR = 1.821(1.324–2.506)], and hsa-miR-365a-3p [p = 0.0004, HR = 1.823(1.309–2.538)] were still correlated with the overall survival of KIRC. And the expression of hsa-miR-21-5p (logFC = 2.167, p < 0.001), hsa-miR-223-3p (logFC = 1.000, p < 0.001), and hsa-miR-365a-3p (logFC = 0.998, p < 0.001) were increased significantly. The patient with low risk score actually exhibited a better overall survival (Fig. 5a). The AUC of the DEMs signature in 1-year was 0.5826 (Fig. 5b). And the time-dependent ROC curves in 3-year and 5-year had AUC values higher than 0.6, which were 0.6016 and 0.6541 respectively (Fig. 5c, d).

Construction of survival risk score system based on DEMs signature. a Survival curves of prognostic model based on DEMs in KIRC. b–d The ROC curve of prognostic model based on DEMs in 1-, 3-, and 5-year with AUC value
Discussion
Previous study indicated that 2.2% of all new cancer cases and 1.8% of all cancer related death was RCC in 2018 globally [1]. And almost 30% of KIRC, the main type of RCC, with localized cancers eventually develop to metastases despite after surgical treatment [4]. Therefore, it is necessary to identify DEGs as suitable prognosis biomarkers for the diagnosis of KIRC. The expression of mRNA was affected by many factors, such as miRNA, lncRNA, methylation, and so on. In the present study, we just focus on the miRNAs. MiRNAs, a class of small noncoding RNAs of ∼22nt in length, are firstly identified in 1993 in Caenorhabditis eleganss. Increasing scientific reports demonstrate that miRNAs are involved in the regulation of almost all the biological phenomena in various species which repress the target transcripts through partial complementarity [5]. Abnormal expression of miRNAs is closely related to the pathogenesis of most human diseases, including cancer [8, 10]. Previous studies have shown that miRNAs are involved in the initiation, progression, and prognosis of various cancers. For instance, Zhang et al. found that miR-1246 and miR-1290 are critical for the tumor initiation and progression of lung cancer [11]. Wang et al. found that miR-200c targets CDK2 and suppresses tumorigenesis in renal cell carcinoma [12]. Therefore, the aim of this study was to find the biomarkers related correlated to abnormal miRNA expression, which has an important role in the diagnosis and prognosis for various cancers. In the present study, we found 20 DEMs and 252 DEGs through correlation analysis. And then, we found 20 KEGG pathways were enriched by functional enrichment analysis, of which 9 signaling pathway was enriched significantly. The other 11 signaling pathways are not significantly enriched, but previous studies also indicated that they also play an important role in the pathogenesis of cancers, such as metabolic related pathways [13,14,15].
Subsequently, we identified that TIMP3 and HMGCS1 were correlated with the overall survival of KIRC patients through bioinformatics analysis. And the constructed prognosis model based on TIMP3 and HMGCS1 could accurately predict the overall survival rate of KIRC patients. TIMP3 (Tissue inhibitor of metalloproteinases-3) belongs to a family of negative regulators of matrix metalloproteinase activity. Previous studies indicated that TIMP3 as a tumor suppressor could modulate tumor migration, invasion, and tumorigenicity [16]. High expression of TIMP3 could promote apoptosis in various tumors [16]. Das et al. found that reduced expression of TIMP3 was observed in 74% of the human malignant melanoma cases [17]. Loss or down regulation of TIMP3 could promote the metastasis, cell growth and invasion of several cancers [18,19,20]. Moreover, Gu et al. and Mylona et al. found that TIMP3 could predict the overall survival rate for hepatocellular carcinoma and breast cancer [21, 22]. In the present study, we also found that the expression of TIMP3 was decreased significantly in KIRC. And the survival analysis also indicated that TIMP3 was correlated with the overall survival rate of KIRC patients which further reinforce the relationship of TIMP with cancers. The KIRC patients with low expression of TIMP3 displayed worse overall survival rate. All of these results reinforced the relationship of TIMP3 with cancers. By retrospective analysis, we found hsa-miR-21-5p was also correlated with overall survival which could be a potential prognosis biomarker for KIRC. Park et al. found hsa-miR-21-5p was more highly expressed in the recurrence group than in the nonrecurrence group of gastric cancer [23]. Chang et al. found that hsa-miR-21-5p may exert protective phenotypes by targeting breast oncogenes that contribute to patient survival [24].
HMGCS1 (3-hydroxy-3-methylglutaryl-CoA synthase 1) is a potential regulatory node in the mevalonate pathway, whose up-regulation is a common transcriptional event in cancer stem cell enriched subpopulations of breast cancer cell lines [25]. Wang et al. also found the expression of HMGCS1 is increased significantly in stomach adenocarcinoma samples of patients and tumorspheres of gastric cancer cells [26]. Additionally, previous reports demonstrated that krüppel-like factors (KLFs) could regulate the expression of various substrates. Yao et al. found that KLF13 was downregulated in colorectal cancer tissues and colorectal cancer cell line [27]. KLF13 knockdown could effectively promote cell proliferation and colony formation. Opposite results were observed in KLF13 overexpressed cells [27]. In the further research, Yao et al. found that KLF13 transcriptionally inhibited HMGCS1 and knockdown of HMGCS1 could suppress the proliferation of colorectal cancer [27]. But in the present study, we found that the expression of HMGCS1 was decreased significantly in KIRC. And by retrospective analysis, we found HMGCS1 correlated miRNAs hsa-miR-223-3p and hsa-miR-365a-3p were correlated with the overall survival of KIRC. The expression of hsa-miR-223-3p and hsa-miR-365a-3p were increased significantly in KIRC. Previous studies indicated that hsa-miR-223-3p and hsa-miR-365a-3p could promote proliferation, migration and invasion of cancer cells [28,29,30,31]. But what very interesting is that previous studies also indicated that hsa-miR-223-3p and hsa-miR-365a-3p could inhibit the invasion and migration of cancer cells [32,33,34,35]. All of those results suggested that the same gene or miRNA may play different roles in different cancers.
Conclusion
In the present study, we found 2 DEGs and 3 DEMs could be the candidate prognosis biomarkers for KIRC patients. The constructed risk models based on 2 DEGs and 3 DEMs could accurately predict the outcome. We just provided an analysis direction depended on theoretical knowledge and clinical outcomes, more scientific research, especially clinical studies, were needed to confirm our findings.
Availability of data and materials
The datasets analysed during the current study are available in TCGA repository (https://portal.gdc.cancer.gov/projects/TCGA-KIRC), under the accession code: Kidney Renal Clear Cell Carcinoma (KIRC).
Abbreviations
- KIRC:
-
Kidney renal clear cell carcinoma
- DEGs:
-
Differential expression genes
- DEMs:
-
Differential expression miRNAs
References
Bray F, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424. https://doi.org/10.3322/caac.21492.
Hu F, Zeng W, Liu X. A gene signature of survival prediction for kidney renal cell carcinoma by multi-omic data analysis. Int J Mol Sci. 2019;20:5720. https://doi.org/10.3390/ijms20225720.
Yang W, et al. Identification of genes and pathways involved in kidney renal clear cell carcinoma. BMC Bioinform. 2014;15 Suppl 17:S2. https://doi.org/10.1186/1471-2105-15-S17-S2.
Hsieh JJ, et al. Renal cell carcinoma. Nat Rev Dis Primers. 2017;3:17009. https://doi.org/10.1038/nrdp.2017.9.
Khan AQ, et al. Role of miRNA-regulated cancer stem cells in the pathogenesis of human malignancies. Cells. 2019;8:840. https://doi.org/10.3390/cells8080840.
Fernandez-Valdivia R, et al. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development. 2011;138:1925–34. https://doi.org/10.1242/dev.060020.
Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–11. https://doi.org/10.1101/gad.291004.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–66. https://doi.org/10.1038/nrc1997.
Fan CN, Ma L, Liu N. Systematic analysis of lncRNA-miRNA-mRNA competing endogenous RNA network identifies four-lncRNA signature as a prognostic biomarker for breast cancer. J Transl Med. 2018;16:264. https://doi.org/10.1186/s12967-018-1640-2.
Paul P, et al. Interplay between miRNAs and human diseases. J Cell Physiol. 2018;233:2007–18. https://doi.org/10.1002/jcp.25854.
Zhang WC, et al. Tumour-initiating cell-specific miR-1246 and miR-1290 expression converge to promote non-small cell lung cancer progression. Nat Commun. 2016;7:11702. https://doi.org/10.1038/ncomms11702.
Huang CC, et al. Garcinol downregulates Notch1 signaling via modulating miR-200c and suppresses oncogenic properties of PANC-1 cancer stem-like cells. Biotechnol Appl Biochem. 2017;64:165–73. https://doi.org/10.1002/bab.1446.
Zaravinos A, et al. New miRNA profiles accurately distinguish renal cell carcinomas and upper tract urothelial carcinomas from the normal kidney. PLoS ONE. 2014;9:e91646. https://doi.org/10.1371/journal.pone.0091646.
Zaravinos A, Deltas C. ccRCC is fundamentally a metabolic disorder. Cell Cycle. 2014;13:2481–2. https://doi.org/10.4161/15384101.2014.947225.
Zaravinos A, et al. Altered metabolic pathways in clear cell renal cell carcinoma: a meta-analysis and validation study focused on the deregulated genes and their associated networks. Oncoscience. 2014;1:117–31. https://doi.org/10.18632/oncoscience.13.
Huang HL, et al. TIMP3 expression associates with prognosis in colorectal cancer and its novel arylsulfonamide inducer, MPT0B390, inhibits tumor growth, metastasis and angiogenesis. Theranostics. 2019;9:6676–89. https://doi.org/10.7150/thno.34020.
Das AM, et al. Association of TIMP3 expression with vessel density, macrophage infiltration and prognosis in human malignant melanoma. Eur J Cancer. 2016;53:135–43. https://doi.org/10.1016/j.ejca.2015.09.014.
Su CW, et al. Loss of TIMP3 by promoter methylation of Sp1 binding site promotes oral cancer metastasis. Cell Death Dis. 2019;10:793. https://doi.org/10.1038/s41419-019-2016-0.
Chen J, Gu Y, Shen W. MicroRNA-21 functions as an oncogene and promotes cell proliferation and invasion via TIMP3 in renal cancer. Eur Rev Med Pharmacol Sci. 2017;21:4566–76.
Wang X, et al. Upregulation of miR-191 promotes cell growth and invasion via targeting TIMP3 in prostate cancer. J BUON. 2018;23:444–52.
Gu X, et al. TIMP-3 expression associates with malignant behaviors and predicts favorable survival in HCC. PLoS ONE. 2014;9:e106161. https://doi.org/10.1371/journal.pone.0106161.
Mylona E, et al. Expression of tissue inhibitor of matrix metalloproteinases (TIMP)-3 protein in invasive breast carcinoma: relation to tumor phenotype and clinical outcome. Breast Cancer Res. 2006;8:R57. https://doi.org/10.1186/bcr1607.
Park SK, et al. MiR 21–5p as a predictor of recurrence in young gastric cancer patients. J Gastroenterol Hepatol. 2016;31:1429–35. https://doi.org/10.1111/jgh.13300.
Chang JT, Wang F, Chapin W, Huang RS. Identification of MicroRNAs as breast cancer prognosis markers through the cancer genome atlas. PLoS ONE. 2016;11:e0168284. https://doi.org/10.1371/journal.pone.0168284.
Walsh CA, et al. The mevalonate precursor enzyme HMGCS1 is a novel marker and key mediator of cancer stem cell enrichment in luminal and basal models of breast cancer. PLoS ONE. 2020;15:e0236187. https://doi.org/10.1371/journal.pone.0236187.
Wang IH, et al. Mevalonate pathway enzyme HMGCS1 contributes to gastric cancer progression. Cancers (Basel). 2020;12:1088. https://doi.org/10.3390/cancers12051088.
Yao W, Jiao Y, Zhou Y, Luo X. KLF13 suppresses the proliferation and growth of colorectal cancer cells through transcriptionally inhibiting HMGCS1-mediated cholesterol biosynthesis. Cell Biosci. 2020;10:76. https://doi.org/10.1186/s13578-020-00440-0.
Liu C, et al. Upregulated lncRNA ADAMTS9-AS2 suppresses progression of lung cancer through inhibition of miR-223-3p and promotion of TGFBR3. IUBMB Life. 2018;70:536–46. https://doi.org/10.1002/iub.1752.
Han LL, et al. MiR-223-3p promotes the growth and invasion of neuroblastoma cell via targeting FOXO1. Eur Rev Med Pharmacol Sci. 2019;23:8984–90. https://doi.org/10.26355/eurrev_201910_19298.
Wang Y, et al. MicroRNA-365 promotes lung carcinogenesis by downregulating the USP33/SLIT2/ROBO1 signalling pathway. Cancer Cell Int. 2018;18:64. https://doi.org/10.1186/s12935-018-0563-6.
Geng J, et al. MicroRNA-365a-3p promotes tumor growth and metastasis in laryngeal squamous cell carcinoma. Oncol Rep. 2016;35:2017–26. https://doi.org/10.3892/or.2016.4617.
Wang X, Tong Z, Liu H. MiR-223-3p targeting epithelial cell transforming sequence 2 oncogene inhibits the activity, apoptosis, invasion and migration of MDA-MB-468 breast cancer cells. Onco Targets Ther. 2019;12:7675–84. https://doi.org/10.2147/OTT.S217019.
Ji Q, et al. miR-223-3p inhibits human osteosarcoma metastasis and progression by directly targeting CDH6. Mol Ther. 2018;26:1299–312. https://doi.org/10.1016/j.ymthe.2018.03.009.
Li J, Shen N, Bai GP, Huang XS. MiR-365a-3p suppresses proliferation and invasion of Hep-2 cells through targeting ten-eleven translocation 1 (TET1). Neoplasma. 2018;65:730–5. https://doi.org/10.4149/neo_2018_171119N752.
Hong YG, et al. miR-365a-3p regulates ADAM10-JAK-STAT signaling to suppress the growth and metastasis of colorectal cancer cells. J Cancer. 2020;11:3634–44. https://doi.org/10.7150/jca.42731.
Acknowledgements
Not applicable.
Funding
This project is financially supported by the Hunan University of Medicine Foundation (2020122004) and Hunan Provincial Science & Technology Department (2020SK51202) to X.X., Hunan Provincial Education Department (20B417, 20C1328) to Z.Y and T.Z. The funding bodies provided financial support but had no other role in the design of the study, data collection, analysis, and interpretation of data, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
X.X., conceived and designed the experiments; M.H., T.Z., and Z.Y., performed the analysis; C. X., Q.W., and Y.L., helped to analyze the data; X.X., wrote the paper. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table S1.
GO function enrichment analysis of the DEGs. Supplementary Table 2. KEGG pathway enrichment analysis of the DEGs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, M., Zhang, T., Yao, ZY. et al. MicroRNA related prognosis biomarkers from high throughput sequencing data of kidney renal clear cell carcinoma. BMC Med Genomics 14, 72 (2021). https://doi.org/10.1186/s12920-021-00932-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-00932-z