
Abstract
Background
In the clinical setting, workflows for analyzing individual genomics data should be both comprehensive and convenient for clinical interpretation. In an effort for comprehensiveness and practicality, we attempted to create a clinical individual whole exome sequencing (WES) analysis workflow, allowing identification of genomic alterations and presentation of neurooncologically-relevant findings.
Methods
The analysis workflow detects germline and somatic variants and presents: (1) germline variants, (2) somatic short variants, (3) tumor mutational burden (TMB), (4) microsatellite instability (MSI), (5) somatic copy number alterations (SCNA), (6) SCNA burden, (7) loss of heterozygosity, (8) genes with double-hit, (9) mutational signatures, and (10) pathway enrichment analyses. Using the workflow, 58 WES analyses from matched blood and tumor samples of 52 patients were analyzed: 47 primary and 11 recurrent diffuse gliomas.
Results
The median mean read depths were 199.88 for tumor and 110.955 for normal samples. For germline variants, a median of 22 (14–33) variants per patient was reported. There was a median of 6 (0–590) reported somatic short variants per tumor. A median of 19 (0–94) broad SCNAs and a median of 6 (0–12) gene-level SCNAs were reported per tumor. The gene with the most frequent somatic short variants was TP53 (41.38%). The most frequent chromosome-/arm-level SCNA events were chr7 amplification, chr22q loss, and chr10 loss. TMB in primary gliomas were significantly lower than in recurrent tumors (p = 0.002). MSI incidence was low (6.9%).
Conclusions
We demonstrate that WES can be practically and efficiently utilized for clinical analysis of individual brain tumors. The results display that NOTATES produces clinically relevant results in a concise but exhaustive manner.
Similar content being viewed by others

Background
Next-generation sequencing (NGS) has proven remarkably beneficial in not only understanding cancer biology but also guiding cancer care [1,2,3]. Various NGS methods are routinely used in cancer care [4, 5]. Targeted sequencing panels, whole-exome sequencing (WES), and whole-genome sequencing (WGS) are the most commonly utilized methods, each with its advantages and limitations [6,7,8]. Targeted sequencing panels are tailored to investigate curated cancer-related information, provide excellent depth, and are suited for working with formalin-fixed paraffin-embedded (FFPE) samples [9, 10]. In contrast, WES/WGS provides more comprehensive genomics data suited for both screening previously investigated/reported variants and exploring novel relevant variants. More comprehensive genomics data also provide additional information such as direct measurement of the mutational burden [11, 12] and exploration of signatures of mutational processes [13, 14]. Brain tumors have complex genetic landscapes [15,16,17]. Therefore, it is beneficial to gather the most comprehensive genomics information for each neurooncology patient. We hence advocate utilizing WES for neurooncological genomics analyses as it gathers comprehensive information with a lower cost than WGS and is technically less challenging to analyze and interpret.
The bioinformatics workflows for variant calling are well established but the clinical interpretation of the identified variants constitutes a bottleneck in the analysis [18]. In the clinical setting, the analysis workflow should produce results that are both exhaustive and suitable for clinical interpretation. Intending to be simultaneously comprehensive and practical, we created a clinical WES workflow tailored for neurooncology. This approach sequentially filters and presents layers of findings relevant to neurooncology (the layers being alterations that are detected in curated collections of clinically-relevant genes). This sequential filtering approach prioritizes highly relevant findings while still reporting less relevant but possibly important findings. This article presents our approach and provides results of the analysis of our findings on a sizable glioma cohort, demonstrating that our approach yields clinically relevant results.
Methods
Reads-to-variants workflow
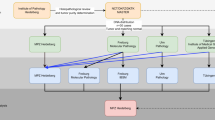
The overview of the complete workflow is presented in Fig. 1. The reads-to-variants pipeline is presented below.

Outline of the reads-to-personalized-report workflow
For quality control, FASTQC (v0.11.9) [19] is used. For tumor and normal samples, the reads are mapped to the reference (hg38) using bwa (version 0.7.17-r1188) [20] and pre-processed, including cleaning the SAM file, sorting SAM by coordinate and converting to BAM, fixing mate information, and marking PCR duplicates (all via Picard version 2.23.8) [21]. For samples that were sequenced in multiple lanes, data for all lanes are combined at this step. Finally, base quality score recalibration (GATK [22] v4.1.9.0) is performed. For quality control, GATK3–DepthOfCoverage (version 3.8-1-0-gf15c1c3ef) and Picard-CollectAlignmentSummaryMetrics are used.
For detecting germline variants (single nucleotide variants (SNVs) and short insertion/deletions (indels)), GATK–HaplotypeCaller is used. For detecting somatic SNV/indels, GATK–MuTect2 is used. Both germline and somatic SNV/indels are annotated using GATK–Funcotator. For detecting somatic copy number alterations (SCNAs), ExomeCNV is used [23]. Annotations of gene-level SCNAs and cytoband annotations are performed via an in-house script.
Personalized neurooncology report workflow
To produce comprehensive reports of WES results, we developed the reporting workflow NOTATES. NOTATES uses curated datasets of glioma- and cancer-related variants and genes to sequentially report clinically relevant findings.
After a summary of somatic WES findings, the report contains the following sections:
-
1
Quality Metrics
-
a.
Summary Table of Quality Metrics
-
b.
Tumor Purity
-
a.
-
2
Germline Alterations
-
a.
ACMG Incidental Findings
-
b.
Variations in Cancer Gene Census Genes
-
c.
Variations in Cancer Predisposition Genes
-
d.
Variations in DNA Damage Repair Genes
-
e.
Common Variants
-
a.
-
3
Somatic Single Nucleotide Variations (SNVs) and Small Insertion/Deletions (Indels)
-
a.
Tumor Mutational Burden (TMB)
-
b.
Microsatellite Instability Status (MSI)
-
c.
Variants in Established Glioma Genes
-
d.
Hotspot Variants in Cancer Gene Census Genes
-
e.
Other Variants in Cancer Gene Census Genes
-
f.
Other Possibly Important Somatic SNV/indels
-
i.
Variants in DNA Damage Repair Genes
-
ii.
Variants in Important KEGG Pathway Genes
-
i.
-
a.
-
4
Somatic Copy Number Alterations (SCNAs)
-
a.
SCNA Burden
-
b.
Established SCNAs in Glioma
-
c.
SCNAs in Cancer Gene Census Genes
-
d.
Broad SCNAs
-
e.
Plots of SCNA Segments by Chromosome
-
a.
-
5
Loss of Heterozygosity (LOH) Events
-
a.
LOH Overview
-
b.
LOH + Somatic SNV/Indel
-
c.
LOH Events in Cancer Gene Census Genes
-
a.
-
6
Genes with Double Hit
-
7
Tumor Heterogeneity Analysis
-
8
Mutational Signatures
-
9
pathfindR—KEGG Pathway Enrichment Analysis
The contents of these sections are detailed in the Results section. NOTATES was written in R [24] and R Markdown.
Analyses and patients
Using NOTATES v1.5, 58 WES analyses from matched blood and tumor samples of 52 patients were analyzed: 47 primary and 11 recurrent diffuse gliomas. Overall, 47 grade IV (81.03%), 7 grade III (12.07%), and 4 grade II tumors (6.9%) were analyzed. Clinical details for all patients and analyses are presented in Additional file 2: Table S1. For each tumor specimen submitted for WES, sections were reviewed by a neuro-pathologist to confirm the diagnosis of diffuse glioma and specifically excise a region within the tumor sample containing only tumor tissue. DNA was extracted using the DNeasy Blood & Tissue Kit (QIAGEN).
All analyses of NOTATES results presented here were performed using R. Selected results were compared with results from the TCGA pan-glioma cohort [15].
Software availability
The reads-to-variants and reporting workflow NOTATES is available for non-commercial purposes on GitHub: https://github.com/egeulgen/NOTATES.
Results
Analysis and reporting of exomes
Sequencing quality metrics
The median mean read depths were 199.88 for tumor and 110.955 for normal samples. The median percentages of reads with at least 25X coverage were 99.3% and 98.55% for tumor and normal samples, respectively. Detailed quality metrics are presented in Additional file 2: Table S2.
Germline variants
Raw germline variants (median = 80,328, range = 72,008–120,635 per patient) are initially filtered according to GATK's best practices [22] for eliminating technical artifacts to yield a median of 64,815 (range = 58,528–87,619) variants per patient (Fig. 2a). For reporting, we only include variants that:
-
have MAF < 1%
-
are not reported as “benign” or “likely benign” in ClinVar [25]
-
have non-synonymous impact
-
are not in FLAGS [26] genes.

Variant analysis and reporting workflow for a germline variants, b somatic short variants, and c somatic copy number alterations
This filtering results in a median of 464 (range = 400–536) variants per patient. A median of 22 (range = 14–33) variants per patient is in the reported categories: A median of 2 (range = 0–6) in “ACMG Incidental Findings”, 16 (range = 10–27) “Variants in Cancer Gene Census Genes”, 0 (range = 0–2) in “Variants in Cancer Predisposition Genes” and 3 (range = 0–7) in “Variants in DNA Damage Repair Genes”.
Considerable percentages of combined reported variants (in all patients) per each category did not have a record in ClinVar (“not reported”) and for variants with a ClinVar record. The most frequent clinical significances were “Drug response” for “ACMG Incidental Findings” (37.3%), “Conflicting” for “Variants in Cancer Gene Census Genes” (5.17%), and “VUS” for “Variants in Cancer Predisposition Genes” (16.67%) and “Variants in DNA Damage Repair Genes” (3.82%) (Additional file 2: Fig. S1). Very small fractions of reported variants per each category were reported as “Pathogenic” or “Likely Pathogenic”: 2.38% for “ACMG Incidental Findings”, 0.48% for “Variants in Cancer Gene Census Genes”, 4.17% for “Variants in Cancer Predisposition Genes” and 1.91% for “Variants in DNA Damage Repair Genes” (Additional file 2: Fig. S1).
Somatic short variants
To filter out sequencing artifacts, raw somatic short variants (median = 14,000, range = 4068–55,533 per analysis) are similarly filtered following the GATK best practices recommendations to result in a median of 223 (range = 57–22,271) variants per analysis (Fig. 2b). For reporting, we further filter these “called” variants and only include variants that:
-
have tumor Variant Allele Frequency (VAF) > 5%
-
have non-synonymous impact
-
are not in FLAGS genes.
This filtering results in a median of 49.5 (range = 2–5646) variants per analysis. A median of 6 (range = 0–590) variants is in the reported categories: A median of 2 (range = 0–44) in “Variants in Established Glioma Genes”, 0 (range = 0–28) in “Hotspot Variants in Cancer Gene Census Genes”, 2 (0–309) in “Other Variants in Cancer Gene Census Genes”, 0 (range = 0–57) in “Variants in DNA Damage Repair Genes” and 1 (range = 0–152) in “Variants in Important KEGG Pathway Genes”.
Figure 3 presents the reasoning behind the sequential filtering of somatic short variants. “Called” (sequencing artifacts excluded) somatic short variants are initially filtered according to the above-mentioned criteria, excluding an average of 78.08% (SD = 8.36%) of “called” variants (Fig. 3a). An average of 2.91% (SD = 1.62%) of “called” variants were reported sequentially in the (1) “Glioma-related” subsection (“Variants in Established Glioma Genes”), (2) “Cancer-related” subsections (“Hotspot Variants in Cancer Gene Census Genes” and “Other Variants in Cancer Gene Census Gene”) and (3) “Selected Gene Sets” subsections (“Variants in DNA Damage Repair Genes” and “Variants in Important KEGG Pathway Genes”). On average, 19.01% (SD = 7.63%) did pass the reporting filter but were not reported. By sequential filtering, a variant reported in a category is not reported in the following ones. A mean percentage of 31.28% (SD = 22.37%) of all reported short somatic variants were in the “Glioma-related” subsection, 42.85% (SD = 21.99%) were in the “Cancer-related” subsections and 25.87% (SD = 22.92%) were in “Selected Gene Sets” subsections (Fig. 3b).

Sequential reporting of called somatic short variants. a Flow diagram displaying the sequential reporting process of “called” (technical artifacts excluded) somatic short variants. Mean (standard deviation) percentages over all “called” somatic short variants are indicated. b Bar plot displaying mean percentage of somatic short variants reported in each category over all reported variants. Error bars indicate standard deviation
Somatic copy number alterations
ExomeCNV analysis yields a median of 3222 (range = 112–42,370) segments per analysis (Fig. 2C). For high confidence, only SCNAs with a |log2(Tumor/Normal) ratio|≥ 0.25 are reported (median = 1964, range = 66–26,636 segments per analysis). For gene SCNA events, under “Established SCNAs in Glioma”, a median of 6 (range = 0–12) SCNA events per analysis are reported, and a median of 0 (range = 0–12) SCNA events per analysis are reported under “SCNAs in Cancer Gene Census Genes”. Under “Broad SCNAs” a median of 19 (range = 0–94) cytoband-level SCNA events per analysis are reported. Chromosomal-arm-level SCNA events in each tumor are presented in Additional file 2: Fig. S2.
Tumor mutational burden and microsatellite instability
The TMB values of all tumors are presented in Additional file 2: Fig. S3A. TMB in primary gliomas (median = 3.2/Mb) were significantly lower than the TMB in recurrent cases (median = 5.8/Mb. Wilcoxon, p = 0.002). The TMB values in different molecular subsets (devised based on WES findings) were also significantly different (Kruskal–Wallis, p = 0.0072. Additional file 2: Fig. S3B).
The TMB distribution of this glioma cohort was comparable to (i.e., not significantly different than) the TMB distributions of the TCGA–Glioblastoma multiforme (GBM) and TCGA-Low-grade Glioma (LGG) cohorts (t-test p = 0.7 and p = 0.37 for GBM and LGG, respectively. Additional file 2: Fig. S3C).
There were 4 cases (6.9%) that were predicted to have microsatellite instability and none of the cases were predicted to have POLE deficiency.
The most frequently reported alterations
Germline variants
The top 10 genes that harbored a reported germline variant in each subsection are presented in Fig. 4a. A large proportion variants reported in the germline variants section had no clinical significance annotation in ClinVar.

Top 10 genes reported in each subsection. a Bar plot displaying the percentages of patients with germline SNV/indel for the top 10 genes in each germline subsection, colored by clinical significance in ClinVar. For patients with multiple germline variants in a given gene, the most severe clinical (as ranked in the legend) significance was used. b Bar plot displaying the percentages of analyses with somatic SNV/indel for the top 10 genes in each somatic short variant subsection in the NOT (dark blue) and the TCGA (light blue) cohorts (top) and boxplots displaying the distributions of variant allele frequencies for the same top 10 genes (bottom), red dashed line indicates the median VAF value of all reported variants. c Bar plot displaying the percentages of analyses with SCNA events in the top 10 genes in “Established SCNAs in Glioma” (left), and top 10 chromosome-/arm-level SCNAs (right), colored by SCNA type (amplification – “Amp” or deletion – “Del”). Selected consensus tumor suppressor genes/oncogenes located within corresponding genomic regions are indicated
The gene with the most frequent reported germline variants under “ACMG Incidental Findings” was TP53 with a single variant rs1042522 (“Drug response” clinical significance for antineoplastic agents response in ClinVar, 92.31% of patients). Under “Variants in Cancer Gene Census Genes”, the genes with the most frequent reported variants were FLT3 with the variant rs1933437 (clinical significance not reported in ClinVar, 73.08%) and XPC with the variants with the variant rs2228001 (“Drug response” clinical significance for cisplatin response—Toxicity/ADR in ClinVar, 73.08%). The gene with most frequent reported germline variants under “Variants in Cancer Predisposition Genes” was COL7A1 with 7 different variants: g.chr3:48569407G>C (not reported, 1.92%), g.chr3:48591687G>T (not reported, 1.92%), rs200868430 (not reported, 1.92%), rs141787797 (not reported, 1.92%), rs116005007 (VUS, 1.92%), rs200505918 (not reported, 1.92%), and rs147633212 (VUS, 1.92%). Under “Variants in DNA Damage Repair Genes”, the DNA polymerase gene POLK was the gene with most frequent germline variants: rs148960463 (Pathogenic, 3.85%), rs368533237 (not reported, 1.92%), rs151251843 (not reported, 1.92%), g.chr5:75596636A>G (not reported, 1.92%), and g.chr5:75581377C>G (not reported, 1.92%).
Under “Common Variants”, the top 5 most frequent single nucleotide polymorphisms (SNPs), previously shown in genome wide association studies (GWASes) to be associated with glioma, were: rs1110784 (in ATP9B, 65.38%), rs1760897 (in TEP1, 48.08%), rs3828550 (in KDR, 11.54%), rs1799782 (in XRCC1, 7.69%) and rs1468358 (in PLOD3, 3.85%).
Somatic short variants
The top 10 genes that harbored a reported somatic short variant per each subsection are presented in Fig. 4b. Overall, there was a positive correlation between the percentages of all the top reported genes between the current cohort (NOT) and the TCGA cohort (Spearman’s ρ = 0.5, p < 0.001). Except for the subsection “Variants in DNA Damage Repair Genes", there were positive correlations of percentages of the top 10 genes between the current cohort (NOT) and the TCGA cohort per each subsection (Fig. 4b, top figure). It can be observed that the most frequent genes were observed in “Variants in Established Glioma Genes”. The gene with the most frequent somatic short variants was TP53 (41.38%).
Figure 4b bottom figure displays the distributions of VAFs of top 10 genes with reported somatic short variants per each somatic short variant subsection. The distributions of tumor VAF values per subsection were significantly different (Kruskal–Wallis test, p < 0.001). The median VAF of “Variants in Established Glioma Genes” was the highest (0.38), followed by “Hotpot Variants in Cancer Gene Census Genes” (0.31), “Other Variants in Cancer Gene Census Genes” (0.28), “Variants in Important KEGG Pathway Genes” (0.22) and “Variants in DNA Damage Repair Genes” (0.19).
Somatic copy number alterations
Under “Established SCNAs in Glioma”, the most frequently reported SCNA events were CDK6 amplification (55.17% of analyses), MET amplification (50%), CDKN2A deletion (44.83%), EZH2 amplification (44.83%), BRAF amplification (43.1%), CDKN2B deletion (43.1%), EGFR amplification (39.66%), RB1 deletion (22.41%), MAX deletion (20.69%), and NF2 deletion (20.69%, Fig. 4c, left).
Chromosomal-arm-level SCNA events in each tumor are presented in Additional file 2: Fig. S2. The most frequently observed chromosome or chromosomal arm level SCNAs were chr7 amplification (46.55%), chr22q loss (34.48%), chr10 loss (27.59%), chr10q loss (25.86%), chr13q loss (24.14%), chr9p loss (22.41%), chr14q loss (18.97%), chr20q amplification (15.52%), chr21p loss (15.52%), and chr6q loss (15.52%, Fig. 4C, right). Frequencies of all SCNA events by cytoband are presented in Additional file 2: Fig. S4, amplifications in chr7 and deletions in chr10 have the highest overall frequencies.
Personalized neurooncology report
To communicate the findings of potential clinical relevance, we developed a comprehensive personalized neurooncology report (Fig. 5, Additional file 1).

Example whole exome sequencing summary report
Summary report of somatic WES findings
The initial page of the report summarizes somatic findings, including TMB, somatic short variants, and somatic copy number alterations on a single page (Fig. 5). The summary includes a description section, providing information on the indication for testing, treatment status, tumor sample type, normal sample type, and DNA extraction method. The summary indicates exome coverage, providing a high-level overview of the quality of the patient's exome data.
For TMB, a plot of all previously analyzed tumor samples’ TMB values, along with the current tumor’s TMB (circled in red) by molecular subset (devised based on WES-identifiable markers) is provided, and the TMB of the current is indicated. The MSI status of the tumor is also indicated in this section.
For somatic short variants, all somatic short variants reported in different categories (i.e., “Variants in Established Glioma Genes”, “Hotspot Variants in Cancer Gene Census Genes”, “Other Variants in Cancer Gene Census Genes”, “Variants in DNA Damage Repair Genes” and “Variants in Important KEGG Pathway Genes”) are presented in a table format, containing information on variant impact classification, protein change annotation, genome change annotation, and tumor VAF.
For SCNAs, a plot of copy number of segments by chromosome is provided as well as a table containing all gene-level SCNAs reported in each category (i.e. “Established SCNAs in Glioma” and “SCNAs in Cancer Gene Census Genes”.
Quality metrics
-
(I)
Summary table of quality metrics
A summary of sequencing quality, including the number of lanes, read type, read length, the total number of reads, PF reads, aligned PF reads, PE aligned, mean coverage, and percentage of bases covered at 1X, 5X, 10X, 25X, 50X and 100X, is reported here.
-
(II)
Tumor purity
The fraction of reads coming from cross-sample contamination, reflecting a measure of tumor purity, is calculated using the GATK-CalculateContamination tool and presented here. A purity/clonality estimate (reflecting normal contamination in the tumor sample) based on copy number alterations is presented under section “Tumor Heterogeneity Analysis”.
Germline alterations
Findings are filtered (except for “Common Variants”) for germline single nucleotide variations (SNVs) and short (typically less than 20 bases-long) insertion-deletion events (indels) that:
-
are not reported as "benign" or "likely benign" in ClinVar [25]
-
have population Minor Allele Frequency (MAF) < 1% (in 1000 Genomes [27], ExAC [28] and ESP6500[29])
-
have non-synonymous impact (one of "Frame_Shift_Del", "Frame_Shift_Ins", "Splice_Site", "Translation_Start_Site","Nonsense_Mutation", "Nonstop_Mutation", "In_Frame_Del","In_Frame_Ins", "Missense_Mutation")
-
are not in genes that are often non-pathogenic and passengers but are frequently mutated in most of the public exome studies (named FLAGS) as collected by Shyr et al. [26]
The section follows a sequential order (except for “Common Variants”) where an alteration reported in a subsection is not reported in the following subsections.
-
a.
ACMG incidental findings
Filtered germline SNV/indels affecting any ACMG SF v2.0 [30] genes for reporting incidental findings are reported here.
-
b.
Variants in cancer gene census genes
This subsection filters the germline SNV/indels for genes that are in the Cancer Gene Census (CGC) from the Catalogue of Somatic Mutations in Cancer (COSMIC), a catalog of genes containing variants associated with cancer [31].
-
c.
Variants in cancer predisposition genes
Genes in which germline variants confer an increased risk of cancer are called cancer predisposition genes. Filtered germline SNV/indels in cancer predisposition genes cataloged by Rahman [32] are reported here.
-
d.
Variants in DNA damage repair genes
Germline variants in genes that take part in the DNA damage repair as collected by the Wood laboratory[33] are reported here.
-
e.
Common variants
Here, germline alterations are filtered for single nucleotide polymorphisms (SNPs) previously shown in genome-wide association studies (GWASes) to have an association with gliomas, as listed in the GWAS catalog [34] under “EFO_0005543” [35].
Somatic SNV/indels
Somatic variants obtained via MuTect2 are filtered to have a variant allele frequency (VAF) of at least 5% and be non-synonymous variants. FLAGS [26] were excluded from the report. The variant subsections in this section also follow a sequential order.
-
a.
Tumor mutational burden
In this subsection, the Tumor Mutational Burden (TMB) is reported. TMB is defined as the number of somatic mutations in the coding region per megabase, including SNVs and indels. This calculation is performed through:
1. keeping variants with VAF > 5% and.
2. keeping variants with a sequence depth > 20X in the tumor and > 10X in the normal.
Two scatter plots and a table summarize the median TMB values overall and for each molecular subset (devised based on WES-identifiable markers) for the current and all previously reported tumors.
-
b.
Microsatellite instability status
The MSI status is predicted using the tool MSIpred [36]. Additionally, polymerase-epsilon deficiency is predicted based on the presence of (a) somatic SNVs/Mb > 60 and (b) somatic indels in single sequence repeats/Mb < 0.18.
The predicted MSI and polymerase-epsilon deficiency statuses are reported here.
-
c.
Variants in established glioma genes
This subsection contains somatic SNV/indels in genes reported in the TCGA pan-glioma study of Ceccarelli et al., which analyzed 1122 WHO grade II-III and IV diffuse-gliomas [15].
-
d.
Hotspot variants in cancer gene census genes
This subsection presents somatic SNV/indels where (a) the gene harboring the variant is listed in CGC and (b) the variant is observed in multiple tumors in COSMIC.
-
e.
Other variants in cancer gene census genes
This subsection lists other somatic variants where the gene harboring the variant is listed in CGC (and the variant is not a hotspot variant).
-
f.
Other possibly important somatic SNV/indels
-
g.
Variants in DNA damage repair genes
Contains possibly important somatic SNV/indels in DNA damage repair genes that are in the list of Human DNA Repair Genes[33]
-
h.
Variants in important KEGG pathway genes
Contains possibly important somatic SNV/indels in selected KEGG [37] pathways (namely “Cell cycle”, “mTOR signaling” and “Pathways in cancer”).
Somatic copy number alterations (SCNAs)
For high confidence, only SCNAs with a |log2(Tumor/Normal) ratio|≥ 0.25 are reported.
-
a.
SCNA Burden
Numerous studies have shown that SCNA burden is an important prognostic marker [38,39,40,41]. In this subsection, 4 metrics of SCNA burden are reported:
-
b.
Total altered length(Mb)
-
c.
Total number of alterations
-
d.
The average length of alterations(kb) = Total altered length/Total number of alterations
-
e.
Weighted Genome Instability Index = estimate of the proportion of the exome with aberrant copy number, weighted on a per chromosome basis
-
f.
Established SCNAs in Glioma
This subsection presents SCNAs that are in a list of gene-level SCNAs curated because of their importance in gliomas (as reported in the aforementioned TCGA pan-glioma study [15]).
-
g.
SCNAs in Cancer Gene Census Genes
This subsection lists SCNAs where the gene subject to copy-number alteration is listed in CGC.
-
h.
Broad SCNAs
This subsection lists SCNA events that span over one or more cytobands.
-
i.
Plots of SCNA Segments by Chromosome
This subsection displays SCNA plots (log2(Tumor/Normal) ratio vs. position) per all chromosomes.
Loss of Heterozygosity (LOH) events
For high confidence, only LOH events for which the absolute difference of B-allele frequencies (|BAFTumor − BAFNormal|) is larger than 0.4 are reported.
-
a.
LOH overview
All LOH events that pass the filter are reported here.
-
b.
LOH + somatic SNV/indel
Here, alterations where a gene has LOH, and a somatic SNV/indel are reported.
-
c.
LOH events in CGC genes
LOH events where the gene subject to LOH is listed in CGC are reported here.
Genes with double hit
A double hit strongly suggests a relevant tumor suppressor gene [42]. In this section, the list of genes with somatic SNV/indel as well as SCNA and/or LOH events are reported.
Tumor heterogeneity analysis
To estimate tumor purity as well as clonal/subclonal SCNAs, THetA [43] is used. In this section, the results of THetA are presented.
Mutational signatures
Somatic mutations in cancer genomes are caused by multiple mutational processes, each of which generates a characteristic mutational signature [14]. Analysis of mutational signatures is becoming routine in clinical cancer genomics as the detected signatures of mutational processes have implications for pathogenesis, classification, prognosis, and even treatment decisions [44, 45].
Using DeConstructSigs [46], the weights of Mutational Signatures v3 (May 2019) from COSMIC [47] are estimated. This section presents the mutational signatures detected in this tumor.
pathfindR—KEGG pathway enrichment analysis
For studying mechanisms underlying oncological processes, KEGG pathway enrichment analyses are performed using the active-subnetwork-oriented enrichment approach of pathfindR [48].
-
a.
Enrichment Results for High-impact Somatic SNV/indels
Genes harboring any somatic non-synonymous variants with a VAF > 5% and not in FLAGS are used for analysis.
-
b.
Enrichment Results for High-impact SCNA
Genes harboring homozygous deletion(Tumor/Normal ratio < 0.5) or multi-copy amplification(Tumor/Normal ratio > 1.5) are used for analysis.
Discussion
The reporting of findings of potential oncological relevance from NGS is rapidly expanding into the clinical area [1,2,3]. In this work, we aimed to present the efficiency and utility of our approach to analyze whole-exome sequencing data of individual gliomas and produce clinically interpretable reports of individual cancer genomes. The approach attempts sequential filtration of various layers of genetic information to assist in clinical decision-making.
It is established that individual tumors may harbor clinically relevant alterations which are not observed frequently in tumors of the same cancer type [49]. In our approach, alterations are prioritized from “highly likely” to “less likely” to be clinically relevant. This is done by sequentially filtering for (1) glioma-related alterations followed by (2) cancer-related alterations followed by (3) alterations in selected gene sets. Through sequential filtering, NOTATES greatly reduces the number of variants to be reported while still retaining the most clinically relevant variants as well as other variants of potential significance.
The clinical interpretation of germline variants in cancer is challenging. The sequential reporting of germline variants in NOTATES allows the clinician to identify any clinically relevant variants. The “ACMG Incidental Findings” section allows the identification of incidental variants, followed by “Variants in Cancer Gene Census Genes” and “Variants in Cancer Predisposition Genes” allowing the identification of cancer-related variants. “Variants in DNA Damage Repair Genes” specifically lists germline variants in DNA damage repair genes, which are important in gliomas because numerous studies have provided evidence that DNA repair deficiency was a central theme in gliomagenesis, a finding also reported in our previous study [50, 51]. Most reported germline variants were not included in ClinVar. As previously reported, the prevalence of “pathogenic” / “likely-pathogenic” germline variants in the ACMG Secondary Findings v2.0 list was low [52] whereas the prevalence of such variants in the cancer-related subsections were relatively higher (among variants with clinical significance annotation in ClinVar).
For somatic SNV/indels, the subsection “Variants in Established Glioma Genes” contains the most likely glioma-specific drivers. Overall, a third of the somatic SNVs reported were in this subsection per tumor. The two following subsections contain somatic variants in CGC genes, pointing to possible oncogenic alterations that are not tumor-type-specific. Hotspot alterations were infrequent but a third of the reported variants per tumor were alterations in CGC genes. The median VAF of the glioma-specific alterations (reported under “Variants in Established Glioma Genes”) was relatively higher than that of alterations reported in the other subsections, emphasizing the importance of this subsection.
For assessing SCNAs, both broad (cytoband-level) and gene-level SCNA events are reported. The most commonly observed (observed in > 25%) chromosomal or arm-level copy-number alterations were chr7 amplification, chr22q deletion, and chr10 deletion, frequently observed alterations in gliomas [53,54,55]. When filtered for SCNAs reported in the TCGA-pan glioma study (presented under “Established SCNAs in Glioma”), each tumor contained a median of 7 such gene-level SCNAs. The most common (observed in > 25%) such SCNA events were CDK6 amplification, MET amplification, BRAF amplification, EZH2 amplification, PTEN deletion, CDKN2A deletion, CDKN2B deletion, EGFR amplification.
TMB and the predicted MSI status, which are both predictive biomarkers for systemic cancer immunotherapy [56,57,58], are included in the report as well. Rather than only providing a hard cut-off value, we provide a plot and a table summarizing the TMB status of all reported gliomas, which enables the clinician to evaluate the TMB status in the relevant context. The TMB distribution of this glioma cohort was similar to those of the TCGA glioma cohorts. As expected, the median TMB value for recurrent tumors was higher than the primary tumors. The TMB values of different glioma molecular subsets were also different. Along with TMB, we also predict MSI status and possible POLE deficiency. As previously reported, the incidence of MSI in diffuse gliomas was low[59,60,61].
Because NOTATES allows the identification of specific genetic alterations indicating differing clinical outcomes in gliomas, the findings in the NOTATES report reflect the severity of the tumor. For example, if a mutation in IDH1/IDH2 is detected, this indicates a better prognosis [62, 63], whereas H3-K27M or G34 mutations imply worse disease outcome [64, 65]. Similarly, IDH-wild-type gliomas with EGFR amplifications and/or chromosome 7 amplifications and chromosome 10 loss can be molecularly defined as GBM, conferring worse prognosis [66, 67]. In addition to specific genetic alterations, NOTATES calculates TMB and evaluates the presence of MSI, further aiding the clinical assessment because these are both predictive biomarkers for systemic cancer immunotherapy [56,57,58].
It is important to emphasize that all findings presented in the NOTATES report complement each other. For example, a high TMB, predicted MSI, somatic variants in mismatch repair genes and mismatch repair deficiency-related mutational signatures will all support highly likely mismatch repair deficiency in a tumor, indicating a higher chance of response to immunotherapy.
Identification of clinically relevant findings from the vast amount of data produced by WES is a substantial challenge [49, 68]. In this work, we aimed to propose a solution to this issue by presenting our approach for reporting of genomic findings from WES data of individual gliomas. Using curated resources, NOTATES investigates and presents various forms of findings of potential clinical importance: germline short variants, somatic short variants, somatic copy-number alterations, loss-of-heterozygosity events, tumor mutational burden, microsatellite instability, and mutational signatures. The NOTATES report is formatted to provide a coherent overview of clinically-relevant genomic findings, enabling the adaptation of WES to the clinical setting. For this purpose, NOTATES utilizes curated sets of relevant genes and databases that collect knowledge about cancer alterations and their relationships to tumor formation and clinical utility and reports the findings in a sequential manner according to clinical relevance. The results in this work demonstrate that NOTATES successfully captures glioma-specific alterations while also reporting possibly relevant cancer-related alterations. The comprehensive report contains the most clinically important findings that may aid in clinical decision-making.
Conclusions
In this work, we presented the outline of and a compilation of results from our WES analysis workflow. The results display that NOTATES produces clinically relevant results in a concise but exhaustive manner. Through this work, we demonstrate that WES can practically and efficiently be adapted to the clinical setting for the analysis of individual gliomas.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. NOTATES is available for non-commercial purposes.
Abbreviations
- NGS:
-
Next generation sequencing
- WES:
-
Whole exome sequencing
- WGS:
-
Whole genome sequencing
- FFPE:
-
Formalin-fixed paraffin-embedded
- SNV:
-
Single nucleotide variant
- indel:
-
Insertion/deletion
- SCNA:
-
Somatic copy number alteration
- MSI:
-
Microsatellite instability
- TMB:
-
Tumor mutational burden
- LOH:
-
Loss of heterozygosity
- VAF:
-
Variant allele frequency
- VUS:
-
Variant of unknown significance
- SD:
-
Standard deviation
- MAF:
-
Minor allele frequency
- CGC:
-
Cancer gene census
- COSMIC:
-
Catalogue of somatic mutations in cancer
- SNP:
-
Single nucleotide polymorphism
- GWAS:
-
Genome-wide Association Study
- LGG:
-
Lower-grade glioma
- GBM:
-
Glioblastoma multiforme
References
Gore M, Larkin J. Precision oncology: where next? Lancet Oncol. 2015;16(16):1593–5.
Senft D, Leiserson MDM, Ruppin E, Ronai ZA. Precision oncology: the road ahead. Trends Mol Med. 2017;23(10):874–98.
Yang HT, Shah RH, Tegay D, Onel K. Precision oncology: lessons learned and challenges for the future. Cancer Manag Res. 2019;11:7525–36.
Nakagawa H, Fujita M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018;109(3):513–22.
Rusch M, Nakitandwe J, Shurtleff S, Newman S, Zhang Z, Edmonson MN, Parker M, Jiao Y, Ma X, Liu Y, et al. Clinical cancer genomic profiling by three-platform sequencing of whole genome, whole exome and transcriptome. Nat Commun. 2018;9(1):3962.
van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30(9):418–26.
Jennings LJ, Arcila ME, Corless C, Kamel-Reid S, Lubin IM, Pfeifer J, Temple-Smolkin RL, Voelkerding KV, Nikiforova MN. Guidelines for Validation of Next-Generation Sequencing-Based Oncology Panels: A Joint Consensus Recommendation of the Association for Molecular Pathology and College of American Pathologists. J Mol Diagn. 2017;19(3):341–65.
Berger MF, Mardis ER. The emerging clinical relevance of genomics in cancer medicine. Nat Rev Clin Oncol. 2018;15(6):353–65.
Kerick M, Isau M, Timmermann B, Sültmann H, Herwig R, Krobitsch S, Schaefer G, Verdorfer I, Bartsch G, Klocker H, et al. Targeted high throughput sequencing in clinical cancer Settings: formaldehyde fixed-paraffin embedded (FFPE) tumor tissues, input amount and tumor heterogeneity. BMC Med Genomics. 2011;4(1):68.
Bewicke-Copley F, Arjun Kumar E, Palladino G, Korfi K, Wang J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput Struct Biotechnol J. 2019;17:1348–59.
Allgauer M, Budczies J, Christopoulos P, Endris V, Lier A, Rempel E, Volckmar AL, Kirchner M, von Winterfeld M, Leichsenring J, et al. Implementing tumor mutational burden (TMB) analysis in routine diagnostics-a primer for molecular pathologists and clinicians. Transl Lung Cancer Res. 2018;7(6):703–15.
Stenzinger A, Allen JD, Maas J, Stewart MD, Merino DM, Wempe MM, Dietel M. Tumor mutational burden standardization initiatives: Recommendations for consistent tumor mutational burden assessment in clinical samples to guide immunotherapy treatment decisions. Genes Chromosomes Cancer. 2019;58(8):578–88.
Bergstrom EN, Huang MN, Mahto U, Barnes M, Stratton MR, Rozen SG, Alexandrov LB. SigProfilerMatrixGenerator: a tool for visualizing and exploring patterns of small mutational events. BMC Genomics. 2019;20(1):685.
Alexandrov LB, Kim J, Haradhvala NJ, Huang MN, Tian Ng AW, Wu Y, Boot A, Covington KR, Gordenin DA, Bergstrom EN, et al. The repertoire of mutational signatures in human cancer. Nature. 2020;578(7793):94–101.
Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell. 2016;164(3):550–63.
Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–7.
Yuzawa S, Nishihara H, Tanaka S. Genetic landscape of meningioma. Brain Tumor Pathol. 2016;33(4):237–47.
Sezerman OU, Ulgen E, Seymen N, Durasi IM: Bioinformatics workflows for genomic variant discovery, interpretation and prioritization. In: Bioinformatics tools for detection and clinical interpretation of genomic variations. Edited by Samadikuchaksaraei A, Seifi M: IntechOpen; 2019.
Babraham Bioinformatics - FastQC A Quality Control tool for High Throughput Sequence Data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Li H: Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint 2013.
Picard Tools - By Broad Institute. http://broadinstitute.github.io/picard/
Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinform. 2013;43:11.10.11-11.10.33.
Sathirapongsasuti JF, Lee H, Horst BAJ, Brunner G, Cochran AJ, Binder S, Quackenbush J, Nelson SF. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27(19):2648–54.
R: The R Project for Statistical Computing. https://www.r-project.org/
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Jang W, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucl Acids Res. 2018;46(D1):D1062-d1067.
Shyr C, Tarailo-Graovac M, Gottlieb M, Lee JJ, van Karnebeek C, Wasserman WW. FLAGS, frequently mutated genes in public exomes. BMC Med Genom. 2014;7:64.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74.
Karczewski KJ, Weisburd B, Thomas B, Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha KE, Cummings BB, et al. The ExAC browser: displaying reference data information from over 60 000 exomes. Nucleic Acids Res. 2017;45(D1):D840-d845.
Exome Variant Server. https://evs.gs.washington.edu/EVS/
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–55.
Sondka Z, Bamford S, Cole CG, Ward SA, Dunham I, Forbes SA. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat Rev Cancer. 2018;18(11):696–705.
Rahman N. Realizing the promise of cancer predisposition genes. Nature. 2014;505(7483):302–8.
Human DNA Repair Genes. https://www.mdanderson.org/documents/Labs/Wood-Laboratory/human-dna-repair-genes.html
Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucl Acids Res. 2019;47(D1):D1005-d1012.
GWAS Catalog. https://www.ebi.ac.uk/gwas/efotraits/EFO_0005543
Wang C, Liang C. MSIpred: a python package for tumor microsatellite instability classification from tumor mutation annotation data using a support vector machine. Sci Rep. 2018;8(1):17546.
Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucl Acids Res. 2017;45(D1):D353-d361.
Zhang L, Feizi N, Chi C, Hu P. Association analysis of somatic copy number alteration burden with breast cancer survival. Front Genet. 2018;9:421.
Hieronymus H, Schultz N, Gopalan A, Carver BS, Chang MT, Xiao Y, Heguy A, Huberman K, Bernstein M, Assel M, et al. Copy number alteration burden predicts prostate cancer relapse. Proc Natl Acad Sci USA. 2014;111(30):11139–44.
Hieronymus H, Murali R, Tin A, Yadav K, Abida W, Moller H, Berney D, Scher H, Carver B, Scardino P et al: Tumor copy number alteration burden is a pan-cancer prognostic factor associated with recurrence and death. Elife 2018, 7.
Mirchia K, Sathe AA, Walker JM, Fudym Y, Galbraith K, Viapiano MS, Corona RJ, Snuderl M, Xing C, Hatanpaa KJ, et al. Total copy number variation as a prognostic factor in adult astrocytoma subtypes. Acta Neuropathologica Commun. 2019;7(1):92.
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–58.
Oesper L, Mahmoody A, Raphael BJ. THetA: inferring intra-tumor heterogeneity from high-throughput DNA sequencing data. Genome Biol. 2013;14(7):R80.
Dong F, Davineni PK, Howitt BE, Beck AH. A BRCA1/2 mutational signature and survival in ovarian high-grade serous carcinoma. Cancer Epidemiol Biomark Prev. 2016;25(11):1511–6.
Secrier M, Li X, de Silva N, Eldridge MD, Contino G, Bornschein J, MacRae S, Grehan N, O’Donovan M, Miremadi A, et al. Mutational signatures in esophageal adenocarcinoma define etiologically distinct subgroups with therapeutic relevance. Nat Genet. 2016;48(10):1131–41.
Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. deconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 2016;17(1):31.
Tate JG, Bamford S, Jubb HC, Sondka Z, Beare DM, Bindal N, Boutselakis H, Cole CG, Creatore C, Dawson E, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucl Acids Res. 2018;47(D1):D941–7.
Ulgen E, Ozisik O, Sezerman OU. pathfindR: An R Package for comprehensive identification of enriched pathways in omics data through active subnetworks. Frontiers in Genetics. 2019;10:858.
Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, Jane-Valbuena J, Friedrich DC, Kryukov G, Carter SL, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20(6):682–8.
Ulgen E, Can O, Bilguvar K, Oktay Y, Akyerli CB, Danyeli AE, Yakicier MC, Sezerman OU, Pamir MN, Ozduman K. Whole exome sequencing-based analysis to identify DNA damage repair deficiency as a major contributor to gliomagenesis in adult diffuse gliomas. J Neurosurg. 2019;132:1–12.
Erasimus H, Gobin M, Niclou S, Van Dyck E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutation Res/Rev Mutation Res. 2016;769:19–35.
Olfson E, Cottrell CE, Davidson NO, Gurnett CA, Heusel JW, Stitziel NO, Chen L-S, Hartz S, Nagarajan R, Saccone NL, et al. Identification of medically actionable secondary findings in the 1000 genomes. PLoS ONE. 2015;10(9):e0135193.
Bigner SH, Mark J, Burger PC, Mahaley MS Jr, Bullard DE, Muhlbaier LH, Bigner DD. Specific chromosomal abnormalities in malignant human gliomas. Cancer Res. 1988;48(2):405–11.
Rey JA, Bello MJ, de Campos JM, Kusak ME, Ramos C, Benitez J. Chromosomal patterns in human malignant astrocytomas. Cancer Genet Cytogenet. 1987;29(2):201–21.
Laigle-Donadey F, Crinière E, Benouaich A, Lesueur E, Mokhtari K, Hoang-Xuan K, Sanson M. Loss of 22q chromosome is related to glioma progression and loss of 10q. J Neurooncol. 2006;76(3):265–8.
Yarchoan M, Hopkins A, Jaffee EM. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N Engl J Med. 2017;377(25):2500–1.
Maleki Vareki S, Garrigos C, Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit Rev Oncol Hematol. 2017;116:116–24.
Chang L, Chang M, Chang HM, Chang F. Microsatellite instability: a predictive biomarker for cancer immunotherapy. Appl Immunohistochem Mol Morphol. 2018;26(2):e15–21.
Leung SY, Chan TL, Chung LP, Chan AS, Fan YW, Hung KN, Kwong WK, Ho JW, Yuen ST. Microsatellite instability and mutation of DNA mismatch repair genes in gliomas. Am J Pathol. 1998;153(4):1181–8.
Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen H-Z, Reeser JW, Yu L, Roychowdhury S. Landscape of microsatellite instability across 39 cancer types. JCO Precision Oncol. 2017;1:1–15.
Martinez R, Schackert HK, Plaschke J, Baretton G, Appelt H, Schackert G. Molecular mechanisms associated with chromosomal and microsatellite instability in sporadic glioblastoma multiforme. Oncology. 2004;66(5):395–403.
Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73.
Han S, Liu Y, Cai SJ, Qian M, Ding J, Larion M, Gilbert MR, Yang C. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br J Cancer. 2020;122(11):1580–9.
Kleinschmidt-DeMasters BK, Levy JMM. H3 K27M-mutant gliomas in adults vs. children share similar histological features and adverse prognosis. Clin Neuropathol. 2018;37(2):53.
Lim KY, Won JK, Park C-K, Kim S-K, Choi SH, Kim T, Yun H, Park S-H. H3 G34-mutant high-grade glioma. Brain Tumor Pathol. 2020;38:4–13.
Brat DJ, Aldape K, Colman H, Holland EC, Louis DN, Jenkins RB, Kleinschmidt-DeMasters B, Perry A, Reifenberger G, Stupp R. cIMPACT-NOW update 3: recommended diagnostic criteria for “Diffuse astrocytic glioma, IDH-wildtype, with molecular features of glioblastoma, WHO grade IV.” Acta Neuropathol. 2018;136(5):805–10.
Stichel D, Ebrahimi A, Reuss D, Schrimpf D, Ono T, Shirahata M, Reifenberger G, Weller M, Hänggi D, Wick W. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018;136(5):793–803.
McLaughlin HM, Ceyhan-Birsoy O, Christensen KD, Kohane IS, Krier J, Lane WJ, Lautenbach D, Lebo MS, Machini K, MacRae CA, et al. A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med Genet. 2014;15(1):134.
Acknowledgements
We are grateful to our patients who endure with courage.
Funding
This research received no external funding.
Author information
Authors and Affiliations
Contributions
Conception and design: KO, OC, EU. Acquisition of data: KO, EU, CAB, SKY, KB. Analysis and interpretation of data: KO, OC, EU, AED. Drafting the article: KO, EU, OC, CAB, AED, MCY, OUS. Critically revising the article: all authors. Reviewed submitted version of manuscript: all authors. Statistical and bioinformatics analyses: EU. Administrative/technical/material support: KO, AED, OUS, NMP. Study supervision: KO. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and informed consent to participate
The study was approved by Acibadem Mehmet Ali Aydinlar University’s institutional review board (ATADEK-2018/7, 17.05.2018). We obtained written informed consent from all subjects before their inclusion in the study. All methods were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
An Example Personalized Neurooncology Report.PDF file containing an example report for a diffuse glioma tumor sample.
Additional file 2.
Supplementary figures and tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ülgen, E., Can, Ö., Bilguvar, K. et al. Sequential filtering for clinically relevant variants as a method for clinical interpretation of whole exome sequencing findings in glioma. BMC Med Genomics 14, 54 (2021). https://doi.org/10.1186/s12920-021-00904-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-021-00904-3