
Abstract
Background
Plasmodium falciparum malaria remains a major health problem in Africa. The mechanisms of pathogenesis are not fully understood. Transcriptomic studies may provide new insights into molecular pathways involved in the severe form of the disease.
Methods
Blood transcriptional levels were assessed in patients with cerebral malaria, non-cerebral malaria, or mild malaria by using microarray technology to look for gene expression profiles associated with clinical status. Multi-way ANOVA was used to extract differentially expressed genes. Network and pathways analyses were used to detect enrichment for biological pathways.
Results
We identified a set of 443 genes that were differentially expressed in the three patient groups after applying a false discovery rate of 10%. Since the cerebral patients displayed a particular transcriptional pattern, we focused our analysis on the differences between cerebral malaria patients and mild malaria patients. We further found 842 differentially expressed genes after applying a false discovery rate of 10%. Unsupervised hierarchical clustering of cerebral malaria-informative genes led to clustering of the cerebral malaria patients. The support vector machine method allowed us to correctly classify five out of six cerebral malaria patients and six of six mild malaria patients. Furthermore, the products of the differentially expressed genes were mapped onto a human protein-protein network. This led to the identification of the proteins with the highest number of interactions, including GSK3B, RELA, and APP. The enrichment analysis of the gene functional annotation indicates that genes involved in immune signalling pathways play a role in the occurrence of cerebral malaria. These include BCR-, TCR-, TLR-, cytokine-, FcεRI-, and FCGR- signalling pathways and natural killer cell cytotoxicity pathways, which are involved in the activation of immune cells. In addition, our results revealed an enrichment of genes involved in Alzheimer’s disease.
Conclusions
In the present study, we examine a set of genes whose expression differed in cerebral malaria patients and mild malaria patients. Moreover, our results provide new insights into the potential effect of the dysregulation of gene expression in immune pathways. Host genetic variation may partly explain such alteration of gene expression. Further studies are required to investigate this in African populations.
Similar content being viewed by others


Background
Plasmodium falciparum malaria remains a leading cause of mortality and morbidity in tropical countries. It encompasses a broad range of clinical phenotypes, including mild and severe forms of the disease. Cerebral malaria (CM), severe anaemia, and respiratory distress are the main syndromes of severe malaria (SM). According to a World Health Organization (WHO) report, major complications account for 429,000 deaths annually [1]. In particular, CM is defined as a diffuse encephalopathy with seizures and impaired consciousness, and its case fatality rate has not changed over decades [2]. Patients are treated with antimalarial drugs, and it is thought that adjunctive therapies, such as anti-inflammatory, vasculo- and neuro-protective therapies, and new biomarkers for early diagnosis are needed. The outcome of infection depends on numerous factors, such as parasite virulence, host age, host immune status, and host and parasite genetics. Genome-wide association studies have been conducted in African populations to identify biomarkers associated with SM and to decipher the molecular basis of the pathogenesis. They have yielded, however, very few significant association results [3,4,5], likely due to a poor knowledge of the linkage disequilibrium patterns in African populations [6], and thus, to a lack of relevant tagSNPs for GWAS in Africa. Whole genome sequencing of individuals living in different African areas should overcome this limitation in the future. The assessment of genome-wide levels of expression is an alternative approach to identify relevant candidate genes and molecular pathways involved in pathogenesis.
In this way, transcriptional studies have been carried out in a CM mouse model, and have led to the discovery of genes, whose up- or down-regulation in several tissues was associated with CM, and which were involved in specific pathways, such as metabolic energy pathways, inflammatory response, antigen presentation, and T cell receptor signalling and pathways related to neurogenesis and neurodegenerative disorders [7, 8]. In humans, transcriptional profiling has been used very recently to search for molecular markers associated with malaria. Transcriptional studies have been carried out either in an in vitro model of CM or on blood cells taken from patients with either SM or mild malaria (MM). The in vitro model study has shown that TNF, platelets, and infected red blood cells significantly influence the expression of genes in endothelial cells [9]. Ex vivo studies that compared gene expression levels in SM patients and those in MM patients identified several host genes whose expression levels were associated with SM in Mali and Malawi [10,11,12], pointed out the involvement of immune pathways, such as interferon pathways and Toll-like receptor signalling [11, 12], and revealed genes involved in neurodegenerative disorders [13]. Moreover, a transcriptional study that investigated gene expression levels in blood taken from CM patients with and without malaria retinopathy revealed that cell adhesion and cytokine pathways were positively associated with malaria retinopathy [10]. In addition, ex vivo parasite transcriptional studies led to the identification of up- or down-regulated parasite genes associated with high parasitaemia or malaria retinopathy, and involved in cell adhesion, glycolysis or DNA replication [14, 15].
For the present study, we conducted a blood transcriptional analysis in Senegalese patients with CM, severe non-cerebral malaria (NCM), or MM in order to identify human genes whose expression levels are associated with the severity of the disease. We identified gene clusters that discriminated between CM and MM patients, and provided evidence of pathways associated with CM.
Methods
Patients and samples
Senegalese patients with clinical malaria were enrolled at the Principal Hospital of Dakar, the National Hospital Centre of Pikine, and the Regional Hospital of Tambacounda. Prior to enrolment, written or verbal information on the study was given in their native language, and informed consent was obtained from the participants or their relatives. At the day of admission, biological data including P. falciparum parasitaemia, haematological and biochemical characteristics were determined by hospitals’ medical laboratories and recorded, as previously described [16]. For all patients, the presence of P. falciparum infection was determined by an immunoassay detecting PfHRP2 (Standard diagnostics-Abott-Inc, Chicago, Illinois, USA). For 12 out of 16 patients P. falciparum parasitaemia was determined by microscopic examination of thin and thick blood smears, prior to anti-malarial treatment. The protocols were approved by the investigator’s institutions, the National Ethical Committee and the Ministry of Health of Senegal. The clinical cases were defined according to the WHO criteria [17]. MM cases were defined on the basis of fever with P. falciparum parasitaemia of < 25,000 parasites/μl of blood, with no evidence of impaired consciousness, convulsions, severe anaemia, hypoglycaemia, respiratory distress, or hypoxia. The presence of a deep coma, lack of purposeful response, lack of response to a painful stimulus by Glasgow score < 9, a diagnosed P. falciparum infection, without other clinically cause of impaired consciousness, such as hypoglycemia, meningitis, and encephalitis according to WHO criteria qualified the patients as having CM. NCM cases were defined on the basis of isolated symptoms of SM such as severe anaemia, hypoglycaemia, respiratory distress, or hypoxia, and the absence of neurological symptoms such as impaired consciousness, convulsions and long-term neurological deficits. Peripheral blood was taken by venipuncture on hospital admission. Blood samples (approximately 5 ml) were diluted and peripheral blood mononuclear cells (PBMC) were collected by Ficoll-Histopaque-1077 (Sigma-Aldrich, Saint-Quentin Fallavier) density-gradient centrifugation for RNA. After washing twice with PBS, PBMCs were stored at − 80 °C.
Microarray processing (RNA extraction, amplification, and hybridization)
RiboNucleic Acid (RNA) extraction was carried out using TRIzol (Gibco, Invitrogen Corporation), according to the manufacturer’s instructions. The quantification of RNA was performed using the NanoDrop ND-1000 UV-Vis spectrophotometer (Thermo Fisher Scientific, Illkirch, France), and the quality of the RNA samples was analysed with the Bioanalyzer 2100 (Agilent Technologies®, Massy, France) and Agilent chips (RNA Nano Chip®). Samples with an RNA Integrity Number (RIN) > 8 were retained. Samples and microarrays were next processed according to Agilent’s recommendations. In brief, total RNA was reverse-transcribed with the AffinityScript RT enzyme and cDNA was transcribed in vitro. cRNA was further obtained using T7 RNA polymerase and labelled using Cy3-CTP, before fragmentation and hybridization (600 ng per sample for 17 h at 65 °C) to a SurePrint G3 Human GE 8x60k microarray. After washing, the microarrays were scanned with the SureScan Agilent Microarray Scanner.
Microarray analysis
The quantification of signals was performed with Agilent’s Feature Extraction Software. From the raw data, data filtering, normalization and data backup were performed using the R statistical software package. The quantile method was used to normalize the data. A filter was applied on the raw data to delete controls, and then a second filter was applied to delete probes that showed a signal under the background; finally, 26,372 probes were further analyzed. Data were deposited in the GEO database under GSE116306. Statistical analysis was performed using the TIGR MeV (MultiExperiment Viewer) v4.1 software (http://mev.tm4.org/#/welcome) and the GeneANOVA program [18, 19]. Figure 1 shows the schematic outline of statistical analyses. Welch t-test was used to compare CM patient gene expression levels with those of MM patients. A multi-way ANOVA was used to take into account the influence of covariates, such as age, gender, and leukocyte counts. The ANOVA model gives an estimation of the contribution of each factor in the total variation of the whole data set. Furthermore, a local ANOVA allows the determination of each contribution for each gene; it gives an estimation of the variation due to the factor studied, and the significance of the estimate. An FDR of 10% was applied to correct for multiple tests. An unsupervised hierarchical clustering of the samples was carried out on the basis of the expression of the selected genes. A ‘1-out-iterative cross-validation’ testing procedure was used to ascertain the accuracy of the classification.

Schematic outline of statistical analyses. Blood gene expression levels in MM, CM, and NCM patients were compared (a), and blood gene expression levels in MM patients were compared to that in CM patients (b). Analyses of variance were carried out to identify significant probes and genes. Enrichment analyses were performed to determine molecular pathways potentially involved in malaria pathogenesis. An FDR of 10% was applied to correct for multiple tests
Functional enrichment analysis
The gene annotation of the selected genes was performed using Genomic Regions Enrichment of Annotation Tool (GREAT), Enrichr, and NetworkAnalyst [20,21,22]. Those programs, which allow a biological interpretation of gene clusters on the basis of Molecular Signatures Database, Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways, Reactome pathways, and protein-protein interactions, were used to assess the functional enrichment for differentially expressed genes. Fisher exact and hypergeometric tests were used, whereas an FDR of 10% was applied to correct for multiple tests.
The selected genes were used to construct and visualize the network using NetworkAnalyst [21]. The underlying protein-protein interactions were obtained from InnateDB [23]. The minimum interaction network option was used, and a first-order interaction network that was composed only of the seed nodes and their direct interactors was created; it was further trimmed to keep only the nodes that connected the seed nodes. The degree centrality and the betweenness centrality were also calculated.
Quantitative real time PCR
The RT-QPCR was performed on all samples from CM and MM patients. Eight genes (ZBTB43, TICAM1, PI4K2A, HIC2, ZNHIT3, NFE2, NCR3 and IL18R1) were selected for validation experiments. RT-PCR was performed for each sample from the total RNA with the VILO SuperScript kit (Invitrogen, Carlsbad, California) based on the protocol provided. In summary, 500 ng of total RNA was mixed with 4 μl of the SuperScrip VILO MasterMix supplied in the kit and 5 pmol of primers in a total volume of 20 μl DEPC-treated water. The samples were vortexed and incubated, respectively, at 25 °C, 10 min and 42 °C, 1 h before the reaction was complete at 85 °C, 5 min. The qPCR testing was performed in 96-well plates with the product of the RT-PCR. Briefly, 2 μl of each sample, diluted to 1/10, was added to a solution containing 5 pmol / μl of each foward and reverse primer (Eurofins Genomics, Ebersberg), 12.5 μL of SYBR Green Master Mix (Thermo Fisher Scientific) and 8.5 μL of RNase / DNase free water. The experiment was carried out with the QuantStudio 6 Flex Real-Time PCR System apparatus using the following steps: pre-incubation cycle at 95 °C during 10 min; 40 amplification cycles at 95 °C during 15 s, and 60 °C during 60 s; 1 melting cycle at 95 °C during 15 s, 60 °C during 1 min, and 95 °C during 15 s. Gene expression levels were then calculated after normalizing the measurements on the GAPDH level and a reference sample.
Results
Patients
Blood samples taken from 16 patients were investigated: 6 from CM patients, 4 from severe NCM patients, and 6 from MM patients. Table 1 shows the patient characteristics in the patient groups. There was no significant difference between the groups for age (P = 0.15), haemoglobin concentration (P = 0.55), red blood cell count (P = 0.87), and leucocyte count (P = 0.81) on the basis of a Kruskal-Wallis test. In contrast, there was a difference for platelet count (P = 0.018).
Gene expression analysis
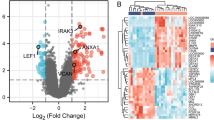
We performed a one-way ANOVA to identify genes whose expression may differ in MM, NCM, and CM patients. Thirty-six probes corresponding to 28 genes showed a difference with a nominal P value threshold of 0.001 (Additional file 1: Table S1), whereas there was no significant difference after applying a false discovery rate (FDR) of 10%. Since the CM patients appeared to show a particular pattern, we carried out a Welch t-test to compare the level of gene expression in CM patients with the level in MM patients. We identified 14 differentially expressed genes on the basis of nominal P values lower than 0.001 (Additional file 1: Table S1). However, after correcting for multiple tests, no significantly expressed genes remained. Figure 2 shows the unsupervised hierarchical clustering based on the 14 genes.

Gene expression profiles in blood samples from cerebral malaria (CM), severe non-cerebral malaria (NCM), and mild malaria (MM) patients. Red and green indicate expression levels above and below the median, respectively. A one-way ANOVA led to the selection of differentially expressed genes in the three patient groups, and those genes were used to carry out a hierarchical clustering (a). A Welch t-test led to the selection of 14 genes, and a hierarchical clustering was performed on this basis (b). Each column represents a patient, whereas each line represents a probe. Selected probes and genes had a nominal P value lower than 0.001. Genes that were down- and up-regulated in CM patients are shown in cluster A and cluster B, respectively. The list of those genes is shown in Additional file 1: Table S1
We further performed a multi-way ANOVA to take into account the effect of potential confounding factors. First, we included MM, NCM, and CM patients, and then we considered only MM and CM patients. Tables 2 and 3 show the result of the global ANOVA; gene factor, gender, age, leukocyte count, and clinical status were sources of variation. Furthermore, we performed an ANOVA for each gene, taking into account the effect of confounding factors. The analysis of the three patient groups identified 503 probes corresponding to 443 genes whose expression levels differed significantly, with an FDR of 10% (Additional file 1: Table S1). For those genes, the median of the variance explained by clinical status was 19.6% (minimum = 4.7% and maximum = 36.2%). On the basis of the expression of those genes, the CM patients differed in their expression compared to MM and NCM patients (Fig. 3). We further investigated the pattern of gene expression in MM and CM patients. After applying an FDR of 10%, we identified 1060 probes corresponding to 842 genes, the expression of which differed in MM and CM (Additional file 1: Table S1). For those genes, the median of the variance explained by CM factor was 40.3% (minimum = 8.7% and maximum = 72.4%). The unsupervised hierarchical clustering of CM-informative genes is presented in Fig. 4. On this basis, the CM patients were clustered together, suggesting that gene expression profiles were well discriminated between cerebral and mild malaria patients. This was further supported by another analysis based on the SVM method. We used a ‘1-out-iterative cross-validation’ testing procedure. Iteratively, 1 out of the 12 samples was removed from the group and was classified based on the correlation between its expression profile and the median profile of samples from CM and MM patients. Accordingly, we correctly classified five out of six cerebral malaria patients and six of six mild malaria patients (P = 0.015).

Hierarchical clustering of cerebral malaria (CM), severe non-cerebral (NCM) and mild malaria (MM) patients on the basis of differentially expressed genes. A multi-way ANOVA, which took into account confounding factors, led to the identification of genes whose expressions differed in CM, NCM, and MM patients after applying an FDR of 10%. Each column represents a patient. Genes that were down- and up-regulated in CM patients are shown in cluster A and cluster B, respectively. The list of those genes is shown in Additional file 1: Table S1

Hierarchical clustering of cerebral malaria (CM) and mild malaria (MM) patients on the basis of differentially expressed genes. A multi-way ANOVA was performed to selectively identify differentially expressed genes in CM and MM patients after an FDR of 10%. Each column represents a patient. Genes that were down- and up-regulated in CM patients are shown in cluster A and cluster B, respectively. The list of those genes is shown in Additional file 1: Table S1
We further analysed the expression of eight selected genes by RT-qPCR to confirm their gene expression patterns. The level of gene expression measured with the microarray method was correlated with that measured with the qPCR method for the studied genes (Additional file 2: Figure S1). The correlation coefficient ranged from 0.69 to 0.94 (P < 0.02). Also, the qPCR results confirmed the microarray results.
Functional enrichment
When analysing the genes showing a differential expressions in CM, NCM, and MM patients, there was no significant functional enrichment on the basis of the Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathways. Nevertheless, the analysis of molecular immune signatures from the Molecular Signatures Database (MSigDB) showed an over-representation of genes regulated in activated CD4 lymphocytes, CD8 lymphocytes, B lymphocytes, NK lymphocytes, macrophages, monocytes, and dendritic cells (Table 4 and Additional file 3: Table S2). Using the differentially expressed genes in the CM and MM groups, we found very similar results (Table 5 and Additional file 3: Table S2).
The products of those genes, whose expressions differed in the MM and SM groups based on the multi-way ANOVA, were further mapped onto a human protein-protein network, leading to subnetworks. Figure 5 shows the protein-protein interaction (PPI) network of significantly regulated genes in MM, CM, and NCM patients, whereas Fig. 6 shows the PPI network of significantly regulated genes in MM and CM patients. The PPI network of genes regulated in MM, CM, and NCM patients was composed of 810 nodes including 322 seed nodes and 2574 edges, whereas the PPI network of genes regulated in MM and CM patients was composed of 1479 nodes including 629 seed nodes and 6479 edges. Additional file 4: Table S3 shows the list of the nodes and their degree and their betweenness centrality measures. When comparing the two PPI networks for the nodes with the highest degrees, the following nodes belong to the best 10 nodes in both networks: UBC, GSK3B, EEF1A1, RELA, APP, and ELAVL1. Noticeably, GSK3B is involved in KEGG pathways, such as TCR- and BCR-signalling and Alzheimer’s disease pathways. RELA is involved in TCR-, BCR-, TLR-, and RIG-I-like receptor-signalling pathways, whereas APP is involved in Alzheimer’s disease pathway. The two PPI networks were significantly enriched for more than 100 Reactome and KEGG pathways (Additional file 5: Table S4). Tables 6 and 7 shows a selection of pathways of interest, such as Alzheimer’s disease, BCR-, TCR-, TLR-, cytokine-, FcεRI-, FCGR-, RIG-I-like receptor-signalling pathways, and natural killer cell mediated cytotoxicity.

Protein-protein interaction (PPI) network of the differentially expressed genes in MM, CM, and CM patients. The size of the nodes is based on their degree centrality, whereas the intensity of their colour reflects their betweenness centrality

Protein-protein interaction (PPI) network of the differentially expressed genes in MM and CM patients. The size of the nodes is based on their degree centrality, whereas the intensity of their colour reflects their betweenness centrality
Discussion
In this study, we investigated blood gene expression patterns in patients with MM, severe NCM, or CM. We identified a molecular signature associated with CM in humans, which was enriched with pathways involved in the activation of immune cells, such as monocytes, macrophages, B and T lymphocytes, and natural killer cells.
We performed microarray experiments and analysed the data using one-way statistical analyses. We identified genes whose expression levels differed at a nominal P value level under 0.001. However, no significant genes remained after multiple test corrections. Since several confounding factors may diminish the power of the analysis, we assessed the effects of age, gender, and leucocyte count on gene expression, and further took their effects into account. Also, we identified 443 genes, the expressions of which differed in MM, severe NCM, and CM patients. This molecular signature mainly discriminated the CM patients from the others; in the same way, severe anaemia and CM patients were reported to differ in their transcriptional and immunological profiles [24, 25]. Furthermore, we focused on CM patients in comparison to MM patients, and identified 842 genes, the expressions of which differed in MM and CM patients. These results are in line with those obtained in blood and brain samples from CM mice [7, 26], and support the existence of blood gene expression profiles associated with CM. Furthermore, they suggest that gene expression profiles might be detected before the occurrence of CM in humans. Such predictive profiles have been identified in mice with CM [7].
Functional enrichment analyses identified several pathways that may be altered by the regulation of gene expression in CM patients. Interestingly, we detected an enrichment of a neurodegenerative disease signature, as previously reported [13, 24], and we identified APP, which is involved in Alzheimer’s disease [27], as a node with a high number of interactions within the PPI network associated with CM. This suggests that some biological pathways are involved in both CM and neurodegenerative diseases. Nevertheless, most of the identified pathways were immune pathways. Strikingly, the functional enrichment of molecular signatures in specific cells using MSigDB data was consistent with the functional enrichment of PPI networks, which were constructed on the basis of regulated genes. These included BCR-, TCR-, TLR-, FcεRI-, FcγR-, and RIG-I-like receptor- signalling pathways, and natural killer cell mediated cytotoxicity pathway. Noticeably, BCR- and TCR- signalling pathways were also associated with CM, on the basis of the brain transcriptional analysis of CM-resistant and CM-susceptible mice [7], whereas the TLR-signalling pathway was associated with SM in Mali [12]. Moreover, there was an over-representation of MSigDB molecular signatures of activated monocytes, dendritic cells, macrophages, NK cells, B lymphocytes, CD8 T lymphocytes, and CD4 T lymphocytes, including Th1, Th2, and T regulatory cells. These findings are consistent with the involvement of those immune cells in malaria pathogenesis. IgG produced by B lymphocytes activate effector cells through FcγRIIA to kill the parasites [28]. Monocytes and macrophages activated through TLR and Th1 effector lymphocytes produce pro-inflammatory cytokines that are thought to be involved in both parasite clearance and immunopathology [29]. Pro-inflammatory cytokines, such as TNF and IFNγ, increase the expression of adhesion molecules on endothelial cells, such as ICAM1, leading to the binding of infected red blood cells on endothelial cells [30,31,32]. It has been suggested that histamine-secreting basophils activated by IgE through FcεRI increase vaso-permeability and the over-expression of ICAM1 on endothelial cells, suggesting that Th2 lymphocytes promoting the production of IgE play a role in malaria [33]. Besides, IFNγ that is produced by Th1 lymphocytes induces the expression of CXCL10 by endothelial cells, leading to an increased adhesion of T lymphocytes [31]. CD8 T lymphocytes recognize plasmodial antigens presented by the MHC class I of the endothelial cells, leading to the death of endothelial cells, to the disruption of the blood-brain barrier, and, potentially, cerebral haemorrhages, as shown in mice with CM [34,35,36]. T regulatory lymphocytes are thought to be the key actors of Th1 effector lymphocyte regulation in malarial infection, and an excessive T regulatory induction, which might lead to high parasitaemia at the early stage of infection, might benefit the host at later stages by preventing an excessive Th1 effector lymphocyte response [37], including an excessive production of pro-inflammatory cytokines. In this way, IL18R and IL1RN that encode for receptors of pro-inflammatory cytokines were up-regulated in malaria patients in Benin [38] and in CM patients in our study population, whereas IL18R was up-regulated in blood from CM mice [26]. Interestingly, IL18 produced by myeloid cells is required for an optimal activation of NK cells upon contact with infected red blood cells [39]. The role of NK cells remains, nevertheless, to be clarified. Although NK cells control malaria infection by killing the parasite and by early producing IFNγ, NK cells may produce inflammatory cytokines in brain microvessels and may amplify the sequestration of infected red blood cells and CD8 T lymphocytes and the disruption of the blood-brain barrier [34, 40]. However, patients with SM were reported to have a lower NK cell cytotoxicity activity than patients with MM, suggesting a protective role against SM [41]. In addition, the RIG-I-like receptor-signalling pathway, which was enriched in genes regulated in CM patients in our study, was shown to be an important pathway to activate the NK cells and kill the parasite [41].
Although our results are in line with published results and propose signal transduction pathways involved in the pathology, they will not provide definitive insights into molecular and cellular mechanisms of cerebral malaria pathogenesis. Also, it is important to point out the limits of the study. First, transcriptional profiling may be influenced by the differences in cellular composition in PBMC. Furthermore, looking for gene expression profiles in well defined cell types will likely provide specific transcriptional profiles associated with CM. Using single-cell RNA sequencing approach would be a promising approach to reach this objective [42]. Second, the sample size of our study is small, and replication studies should be performed. Third, studying blood cells gives an incomplete view of CM pathogenesis mechanisms, because other cells and tissues are involved. In particular, brain microvascular endothelial cells play a central role in CM [30, 31, 43, 44]. However, obtaining brain endothelium in humans is very difficult, and in vitro and in vivo models are rather used to investigate the role of such cells in CM. Fourth, it is not possible to determine the molecular factors causing CM on the basis of our results, because gene expression levels were measured at the time of malaria attacks. Measuring gene expression levels in patients before CM attacks is very challenging. Interestingly, such studies that have been performed in mice infected by Plasmodium berghei ANKA led to identify predictive profiles before the onset of cerebral malaria [7]. This suggests that new candidate causal factors can be identified in this in vivo model and can be tested using gene targeting approach in mice infected by Plasmodium berghei ANKA. It should be stressed, however, that some authors wrote that the murine model is not relevant for the human disease, whereas other authors pointed out that there are many common features between mouse pathology and human pathology [30, 31, 44, 45]. In particular, White et al. maintained that mice with CM show no or little parasite sequestration within the brain, and that human CM mainly results from parasite sequestration and brain microvessels obstruction [46]. Interestingly, Strangward et al. recently showed that the accumulation of parasitized red blood cells in brain microvessels was a feature of mouse CM that was not observed during uncomplicated malaria attacks [47]. It is likely, however, that a single mouse model does not fully reflect the biological mechanisms underlying CM in humans. Noticeably, there is accumulating evidence of the critical role of CD8+ T lymphocytes in mouse CM, whereas there is little evidence of high numbers of CD8+ T lymphocytes in human brain microvessels during CM attacks. Brain endothelial cells have been shown to crosspresent Plasmodium berghei ANKA antigens to mouse CD8+ T lymphocytes [34, 35], whereas it has not been reported in humans. Nevertheless, a human brain endothelial cell line has been shown to phagocytose P. falciparum merozoites, suggesting that human endothelial cells may crosspresent plasmodial antigens to human CD8+ T lymphocytes [35]. Also, we consider that the mouse CM model can provide new functional hypotheses that should be tested in humans.
Conclusions
We identified genes expression profiles that discriminated between CM and MM patients on the one hand, and immune molecular pathways associated with CM on the other hand. Our results are in line with the involvement of monocytes, macrophages, dendritic cells, and B and T lymphocytes, and NK cells in malaria pathogenesis. Further studies are required to confirm the molecular signatures of CM, and to identify those of severe NCM. In addition, further comparisons of severe malaria patient gene expression levels to that of mild malaria patients may reveal a molecular signature common to severe malaria phenotypes. Finally, genetic variation that influences gene expression in patients with clinical malaria may modulate the immunopathological response. We think that looking for such regulatory variants will help to better uncover the genetic basis of severe forms of malaria in African populations.
Availability of data and materials
Data are either included in the published paper or available in the GEO database under GSE116306.
Abbreviations
- CM:
-
Cerebral Malaria
- FDR:
-
False Discovery Rate
- MM:
-
Mild Malaria
- NCM:
-
Non-Cerebral Malaria
- PBMC:
-
Peripheral Blood Mononuclear Cells
- PPI:
-
Protein-Protein Interaction
- RNA:
-
RiboNucleic Acid
- SM:
-
Severe Malaria
- SVM:
-
Support Vector Machine
References
WHO. World Malaria Report 2016. Genova: World Health Organization; 2016.
Manning L, Laman M, Davis WA, Davis TM. Clinical features and outcome in children with severe Plasmodium falciparum malaria: a meta-analysis. PLoS One. 2014;9(2):e86737.
Band G, Le QS, Jostins L, Pirinen M, Kivinen K, Jallow M, Sisay-Joof F, Bojang K, Pinder M, Sirugo G, et al. Imputation-based meta-analysis of severe malaria in three African populations. PLoS Genet. 2013;9(5):e1003509.
Jallow M, Teo YY, Small KS, Rockett KA, Deloukas P, Clark TG, Kivinen K, Bojang KA, Conway DJ, Pinder M, et al. Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet. 2009;41(6):657–65.
Timmann C, Thye T, Vens M, Evans J, May J, Ehmen C, Sievertsen J, Muntau B, Ruge G, Loag W, et al. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature. 2012;489(7416):443–6.
Teo YY, Small KS, Kwiatkowski DP. Methodological challenges of genome-wide association analysis in Africa. Nat Rev Genet. 2010;11(2):149–60.
Delahaye NF, Coltel N, Puthier D, Barbier M, Benech P, Joly F, Iraqi FA, Grau GE, Nguyen C, Rihet P. Gene expression analysis reveals early changes in several molecular pathways in cerebral malaria-susceptible mice versus cerebral malaria-resistant mice. BMC Genomics. 2007;8:452.
Delahaye NF, Coltel N, Puthier D, Flori L, Houlgatte R, Iraqi FA, Nguyen C, Grau GE, Rihet P. Gene-expression profiling discriminates between cerebral malaria (CM)-susceptible mice and CM-resistant mice. J Infect Dis. 2006;193(2):312–21.
Barbier M, Faille D, Loriod B, Textoris J, Camus C, Puthier D, Flori L, Wassmer SC, Victorero G, Alessi MC, et al. Platelets alter gene expression profile in human brain endothelial cells in an in vitro model of cerebral malaria. PLoS One. 2011;6(5):e19651.
Feintuch CM, Saidi A, Seydel K, Chen G, Goldman-Yassen A, Mita-Mendoza NK, Kim RS, Frenette PS, Taylor T, Daily JP. Activated neutrophils are associated with pediatric cerebral malaria vasculopathy in Malawian children. MBio. 2016;7(1):e01300–15.
Krupka M, Seydel K, Feintuch CM, Yee K, Kim R, Lin CY, Calder RB, Petersen C, Taylor T, Daily J. Mild Plasmodium falciparum malaria following an episode of severe malaria is associated with induction of the interferon pathway in Malawian children. Infect Immun. 2012;80(3):1150–5.
Sobota RS, Dara A, Manning JE, Niangaly A, Bailey JA, Kone AK, Thera MA, Djimde AA, Vernet G, Leissner P, et al. Expression of complement and toll-like receptor pathway genes is associated with malaria severity in Mali: a pilot case control study. Malar J. 2016;15:150.
Cabantous S, Doumbo O, Poudiougou B, Louis L, Barry A, Oumar AA, Traore A, Marquet S, Dessein A. Gene expression analysis reveals genes common to cerebral malaria and neurodegenerative disorders. J Infect Dis. 2017;216(6):771–5.
Tonkin-Hill GQ, Trianty L, Noviyanti R, Nguyen HHT, Sebayang BF, Lampah DA, Marfurt J, Cobbold SA, Rambhatla JS, McConville MJ, et al. The Plasmodium falciparum transcriptome in severe malaria reveals altered expression of genes involved in important processes including surface antigen-encoding var genes. PLoS Biol. 2018;16(3):e2004328.
Almelli T, Nuel G, Bischoff E, Aubouy A, Elati M, Wang CW, Dillies MA, Coppee JY, Ayissi GN, Basco LK, et al. Differences in gene transcriptomic pattern of Plasmodium falciparum in children with cerebral malaria and asymptomatic carriers. PLoS One. 2014;9(12):e114401.
Thiam A, Baaklini S, Mbengue B, Nisar S, Diarra M, Marquet S, Fall MM, Sanka M, Thiam F, Diallo RN, et al. NCR3 polymorphism, haematological parameters, and severe malaria in Senegalese patients. PeerJ. 2018;6:e6048.
Severe falciparum malaria. World Health Organization, Communicable Diseases Cluster. Trans R Soc Trop Med Hyg. 2000;94(Suppl 1):S1–90.
Didier G, Brezellec P, Remy E, Henaut A. GeneANOVA--gene expression analysis of variance. Bioinformatics. 2002;18(3):490–1.
Saeed AI, Sharov V, White J, Li J, Liang W, Bhagabati N, Braisted J, Klapa M, Currier T, Thiagarajan M, et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques. 2003;34(2):374–8.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90–7.
Xia J, Benner MJ, Hancock RE. NetworkAnalyst--integrative approaches for protein-protein interaction network analysis and visual exploration. Nucleic Acids Res. 2014;42(Web Server issue):W167–74.
McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28(5):495–501.
Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. InnateDB: systems biology of innate immunity and beyond--recent updates and continuing curation. Nucleic Acids Res. 2013;41(Database issue):D1228–33.
Nallandhighal S, Park GS, Ho YY, Opoka RO, John CC, Tran TM. Whole-blood transcriptional signatures composed of erythropoietic and Nrf2-regulated genes differ between cerebral malaria and severe malarial anemia. J Infect Dis. 219(1):154–64.
Thuma PE, van Dijk J, Bucala R, Debebe Z, Nekhai S, Kuddo T, Nouraie M, Weiss G, Gordeuk VR. Distinct clinical and immunologic profiles in severe malarial anemia and cerebral malaria in Zambia. J Infect Dis. 2011;203(2):211–9.
Oakley MS, Anantharaman V, Venancio TM, Zheng H, Mahajan B, Majam V, McCutchan TF, Myers TG, Aravind L, Kumar S. Molecular correlates of experimental cerebral malaria detectable in whole blood. Infect Immun. 2011;79(3):1244–53.
Wang X, Zhou X, Li G, Zhang Y, Wu Y, Song W. Modifications and trafficking of APP in the pathogenesis of Alzheimer's disease. Front Mol Neurosci. 2017;10:294.
Grant AV, Roussilhon C, Paul R, Sakuntabhai A. The genetic control of immunity to Plasmodium infection. BMC Immunol. 2015;16:14.
Schofield L, Grau GE. Immunological processes in malaria pathogenesis. Nat Rev Immunol. 2005;5(9):722–35.
van der Heyde HC, Nolan J, Combes V, Gramaglia I, Grau GE. A unified hypothesis for the genesis of cerebral malaria: sequestration, inflammation and hemostasis leading to microcirculatory dysfunction. Trends Parasitol. 2006;22(11):503–8.
Ghazanfari N, Mueller SN, Heath WR. Cerebral malaria in mouse and man. Front Immunol. 2018;9:2016.
Riley EM, Stewart VA. Immune mechanisms in malaria: new insights in vaccine development. Nat Med. 2013;19(2):168–78.
Mecheri S. Contribution of allergic inflammatory response to the pathogenesis of malaria disease. Biochim Biophys Acta. 2012;1822(1):49–56.
Howland SW, Claser C, Poh CM, Gun SY, Renia L. Pathogenic CD8+ T cells in experimental cerebral malaria. Semin Immunopathol. 2015;37(3):221–31.
Howland SW, Poh CM, Renia L. Activated brain endothelial cells cross-present malaria antigen. PLoS Pathog. 2015;11(6):e1004963.
Razakandrainibe R, Pelleau S, Grau GE, Jambou R. Antigen presentation by endothelial cells: what role in the pathophysiology of malaria? Trends Parasitol. 2012;28(4):151–60.
Scholzen A, Minigo G, Plebanski M. Heroes or villains? T regulatory cells in malaria infection. Trends Parasitol. 2010;26(1):16–25.
Idaghdour Y, Quinlan J, Goulet JP, Berghout J, Gbeha E, Bruat V, de Malliard T, Grenier JC, Gomez S, Gros P, et al. Evidence for additive and interaction effects of host genotype and infection in malaria. Proc Natl Acad Sci U S A. 2012;109(42):16786–93.
Artavanis-Tsakonas K, Eleme K, McQueen KL, Cheng NW, Parham P, Davis DM, Riley EM. Activation of a subset of human NK cells upon contact with Plasmodium falciparum-infected erythrocytes. J Immunol. 2003;171(10):5396–405.
Wolf AS, Sherratt S, Riley EM. NK cells: uncertain allies against malaria. Front Immunol. 2017;8:212.
Ye W, Chew M, Hou J, Lai F, Leopold SJ, Loo HL, Ghose A, Dutta AK, Chen Q, Ooi EE, et al. Microvesicles from malaria-infected red blood cells activate natural killer cells via MDA5 pathway. PLoS Pathog. 2018;14(10):e1007298.
Ranzoni AM, Strzelecka PM, Cvejic A. Application of single-cell RNA sequencing methodologies in understanding haematopoiesis and immunology. Essays Biochem. 2019;63(2):217–25.
Dunst J, Kamena F, Matuschewski K. Cytokines and chemokines in cerebral malaria pathogenesis. Front Cell Infect Microbiol. 2017;7:324.
Lou J, Lucas R, Grau GE. Pathogenesis of cerebral malaria: recent experimental data and possible applications for humans. Clin Microbiol Rev. 2001;14(4):810–20 table of contents.
Riley EM, Couper KN, Helmby H, Hafalla JC, de Souza JB, Langhorne J, Jarra WB, Zavala F. Neuropathogenesis of human and murine malaria. Trends Parasitol. 2010;26(6):277–8.
White NJ, Turner GD, Medana IM, Dondorp AM, Day NP. The murine cerebral malaria phenomenon. Trends Parasitol. 2010;26(1):11–5.
Strangward P, Haley MJ, Shaw TN, Schwartz JM, Greig R, Mironov A, de Souza JB, Cruickshank SM, Craig AG, Milner DA Jr, et al. A quantitative brain map of experimental cerebral malaria pathology. PLoS Pathog. 2017;13(3):e1006267.
Acknowledgements
The authors would like to thank all the patients who have participated in the study. We thank Aurelie Bergon for managing the deposit of the dataset in the GEO database, and Sandrine Marquet for reading the manuscript.
Funding
This work was supported by the Institute Pasteur of Dakar, the Institute Pasteur of Paris, the French embassy in Senegal, INSERM, and the university of Aix-Marseille. MS was supported by INSERM and PACA regional council. The funding bodies played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
AT, RND, FT, GD and BM performed sample collection and selection. BM, AT and AD collected patient information and were in charge of clinical management. NFN, GV, and AT performed RNA extraction, RNA quality analysis, and microarray experiments. MT and MS performed qPCR validation experiments. AT, CN, AD, MS, RND and PR designed and planned the experiments. MS and PR performed statistical analyses. MS performed network analyses. AT and MS prepared the figures and the tables. PR wrote the manuscript, and AT, MS, RND, BM, CN, GD and AD reviewed the drafts. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocols were approved by the investigator’s institution, the Comité d’Ethique de la Recherche de l’Université Cheikh Anta Diop de Dakar, and the National Ethics Committee for Health Research and Ministry of Health and Social Action of Senegal. A written informed consent was obtained from each participant or their parents or legal guardians for any minors prior to inclusion, after giving them written or verbal information in their native language.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Differentially expressed genes in MM vs CM vs NCM and MM vs CM analyses. Results of the Welch t test, one-way ANOVA analyses, and multi-way ANOVA analyses are shown.
Additional file 2: Figure S1.
The correlation between gene expression levels measured by qPCR and those measured by microarray technology for CM and MM patients. Eight genes were selected. The qPCR data were analysed by the 2-deltadeltaC(t) method with GAPDH as a control gene. Both the qPCR and the microarray data were normalized on the basis of the values obtained with a reference sample.
Additional file 3: Table S2.
Enriched MSigDB signatures for MM vs CM vs NCM and MM vs CM analyses.
Additional file 4: Table S3.
Protein-protein interaction (PPI) network nodes: degree and betweenness centrality for MM vs CM vs NCM and MM vs CM analyses.
Additional file 5: Table S4.
Enriched Reactome and KEGG pathways for MM vs CM vs NCM and MM vs CM analyses.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Thiam, A., Sanka, M., Ndiaye Diallo, R. et al. Gene expression profiling in blood from cerebral malaria patients and mild malaria patients living in Senegal. BMC Med Genomics 12, 148 (2019). https://doi.org/10.1186/s12920-019-0599-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-019-0599-z