
Abstract
Background
Immunoglobulin A nephropathy (IgAN) is the most common primary glomerulonephritis worldwide. Recent evidence suggests that genetic factors are related to the pathogenesis of IgAN. We conducted a genome-wide association study (GWAS) to identify novel genetic susceptibility loci for IgAN in a Korean population.
Methods
We enrolled 188 biopsy-confirmed IgAN cases and 455 healthy controls for the discovery stage and explored associations between IgAN and single nucleotide polymorphisms (SNPs) using a customized DNA chip. The significant SNPs from the discovery samples were then selected for replication in an independent cohort with 310 biopsy-confirmed IgAN cases and 438 healthy controls.
Results
In the first stage, two SNPs (rs10172700 in LOC105373592 and rs2296136 in ANKRD16) were selected for further association analysis in the next stage. In the replication cohort, rs2296136 in ANKRD16 was significantly associated with IgAN (odds ratio [OR] = 1.40, 95% confidence interval [CI] 0.99–1.98, p = 0.05 in log-additive model, OR = 1.55, 95% CI = 1.06–−2.27, p = 0.02 in dominant model, and OR = 0.70, 95% CI = 0.17–−2.84, p = 0.62 in recessive model). rs2296136 in ANKRD16 also showed a significant association with IgAN in the entire study population combining GWAS and replication study (p = 0.0045 in log-additive model, p = 0.0027 in dominant model, and p = 0.76 in recessive model).
Conclusions
The SNPs identified in the present study could be good candidate markers for predicting IgAN in Koreans, although further experimental validation is needed.
Similar content being viewed by others


Background
Immunoglobulin A nephropathy (IgAN) is the most common primary glomerulonephritis worldwide [1]. Its clinical features vary, and it has been recognized as an important cause of kidney failure [1, 2]. The prevalence of IgAN varies substantially according to geographic region [3]. Individuals of Asian descent are more likely to be affected than individuals from other ethnic backgrounds [2]. Familial clustering of IgAN has also been recognized throughout the world [4]. Moreover, some studies have demonstrated immunologic defects and urinary abnormalities in asymptomatic family members of patients with IgAN [4, 5]. Taken together, these findings suggest that genetic factors strongly influence the pathogenesis of IgAN.
In the last two decades, candidate gene association studies and linkage studies seeking candidate genes for IgAN [6, 7] have reported several candidate genes involved in glycosylation, immune regulation, and cytokine pathway. However, these studies have some sample size limitations and methodological problems [7]. Recently, genome-wide association studies (GWASs) have recognized several susceptibility loci for IgAN [8,9,10,11]. GWASs enable the identification of common alleles in complex disease. In contrast to prior studies, GWASs have been shown to identify susceptibility variants even in the setting of significant locus heterogeneity [2]. However, there are some limitations inherent in GWASs. First, GWASs detect only common disease-causing variants that have relatively small effect size. Second, most of the loci are noncoding, and many are located far from the discovered genes. Third, GWASs are not always replicated across studies or populations. Lastly, the previous GWAS DNA chips were fixed and offered less coverage of single nucleotide polymorphisms (SNPs) in exon and promoter regions.
The aim of this study was to identify novel genetic susceptibility loci for IgAN in a Korean population using a customized DNA chip, containing mostly exon and promoter region. We conducted a two-stage GWAS of biopsy-confirmed IgAN with 188 cases and 455 healthy controls in the discovery phase and with independent cohort of 310 cases and 438 healthy controls in the validation phase of the two significant SNPs. To overcome the restrictions inherent in small sample size, we selected biopsy-confirmed IgAN patients from multiple centers.
Method
Study design and subjects
We conducted a two-stage GWAS of IgAN in a Korean population. The first stage (discovery cohort) consisted of a GWAS, and the second stage (validation cohort) was a replication analysis of the top SNP signals that were identified during discovery. Samples with a call rate < 97% or gender mismatch were removed for sample quality control in both the first and second stages. The first stage included 188 patients with biopsy-confirmed IgAN from three kidney centers (Kyung Hee University Medical Center, Seoul, Korea; Kyung Hee University Hospital at Gangdong, Seoul, Korea; and Inje University Busan Paik Hospital, Korea) and 455 healthy controls from the general health check-up program. The second stage included 310 patients with biopsy-confirmed IgAN from the KoreaN Cohort Study for Outcomes in Patients With Chronic Kidney Disease (KNOW-CKD) cohort and 438 healthy controls from the general health check-up program. The KNOW-CKD study is a multicenter, prospective cohort study of adults with CKD in Korea; the study design has been described previously [12]. The healthy controls enrolled in both stages were recruited from the general health check-up program using patients with 1) normal renal function (estimated glomerular filtration rate > 90 mL/min/1.73 m2, and 2) no evidence of kidney injuries in the urine analysis, and 3) no structural problems in the kidney. We calculated the statistical sample power of discovery set using a genetic power calculator (http://osse.bii.a-star.edu.sg/calculation2.php).
All study procedures complied with the ethical guidelines of the 1975 Declaration of Helsinki, as revised in 2000. The study protocol was approved by the Institutional Review Board of all centers, and the approval number was 2012–01-130 obtained from Kyung Hee University Hospital at Gangdong. Written informed consent was obtained from all participants.
Design of the customized DNA chip
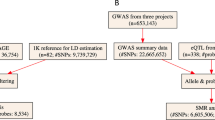
As shown in Fig. 1, we first selected 23,864 Homo sapiens genes from the NCBI gene database (https://www.ncbi.nlm.nih.gov/gene) and manually searched for all known SNPs in all selected genes using the dbSNP database (https://www.ncbi.nlm.nih.gov/snp/). Finally, 241,050 candidate SNPs that were previously reported to be associated with human diseases such as glomerulonephritis, malignancies, autoimmune disorders, and psychiatric disorders were included in this study. We considered regions (exon and promoter regions) for candidate SNPs to surmount the limitation of previous GWAS DNA chips. Candidate SNPs selected according to the following criteria: (1) SNPs located in exon and promoter regions in each gene; (2) SNPs studied in previous GWASs or case and control studies with various diseases; (3) SNPs reported in Asians; (4) SNPs with > 10% minor allele frequency (MAF) in Asian; (5) > 0.1heterozygosity. We then, added 137,657 SNPs from Affymetrix (Affymetrix, CA, USA) GWAS chips which provide high genetic coverage in East Asian Populations. We finally designed a customized chip using the Axiom™ Genome-Wide Human Assay.

Workflow of genetic selection and creation of the customized DNA chip. SNP, single nucleotide polymorphism; GWAS, genome-wide association study; DNA, deoxyribonucleic acid
DNA isolation, genotyping, and quality controls
Genomic DNA was extracted from peripheral blood samples collected in tubes coated with EDTA using a commercially available Roche DNA extraction Kit (Roche, IN, USA). We used the Customer Axiom Exome Array by Affymatrix (Affymatrix, CA, USA) in order to genotype selected SNPs. The experimental process was carried out by Theragen, Suwon, Korea. The following exclusion criteria were applied for SNP quality control: a genotype call rate < 97% and a Hardy–Weinberg equilibrium p-value < 1 × 10− 4 in the controls. Overall, 98,667 SNPs remained after quality control.
Replication analysis
In the second stage, the two significant SNPs identified from the first stage were genotyped. The criterion for candidate SNP selection was the association of a SNP with a p-value ≤5 × 10− 5 in our GWAS. Due to small sample size, there was no genetic association with a p-value less than 1 × 10− 8 that was a best-powered definition for the assessed number of SNPs. Genotyping of new samples from the independent cohort (310 cases and 438 controls) for validation was conducted by direct sequencing after genomic DNA was amplified using specific primers for each gene.
Statistical analysis
In the GWAS analysis, association testing was done with PLINK using logistic regression in order to search candidate SNPs for IgAN in a Korean population (http://pngu.mgh.harvard.edu/~purcell/plink/). The quantile-quantile (Q-Q) and Manhattan plots were calculated using the statistical analysis program R (http://www.r-project.org/). In the replication phase, SNPstats (http://bioinfo.iconcologia.net/index.php) and SPSS 23.0 (SPSS Inc., Chicago, IL, USA) were used to calculate odds ratios (OR), 95% confidence intervals (CI), and p-value. Genetic models [dominant (major homozygous versus. Heterozygous + minor homozygous), recessive (major homozygous + heterozygous versus. Minor homozygous, and log-additive (major homozygous versus. Heterozygous versus. Minor homozygous) models] were applied.
Results
GWAS identifies two IgAN-susceptible SNPs in a Korean population
In the first stage, a GWAS analysis was performed with 188 IgAN cases and 455 healthy controls using 98,667 SNPs. The clinical characteristics of the cases and controls are summarized in Table 1. A Q-Q plot of observed versus expected p-values revealed significant associations between IgAN and certain SNPs (Fig. 2A). As shown in Fig. 2B, there was a significant signal of association with chromosome 6p. Because of the small sample number, there were no significant (p < 1 × 10− 8) gene associations with IgAN. For replication analysis, we excluded SNPs with MAF < 0.05, and selected only one of the most significant SNPs in the same gene. Two SNPs with suggestive evidence for association at p ≤ 5 × 10− 5 were selected for further association analysis (Table 2) in the second stage. The statistical sample power calculated using a genetic power calculator was 79.8% for rs10172700 and 75.1% for rs2296136.

a, Quantile-quantile (Q-Q) plot of p-values for test statistics (Cochran-Armitage trend tests) in the GWAS. b, Manhattan plot showing the -log10P values of 98,667 SNPs in the GWAS for 188 IgAN patients and 455 healthy controls. GWAS, genome-wide association study; SNP, single nucleotide polymorphism; IgAN, Immunoglobulin A nephropathy
Replication study validates association of two SNPs with IgAN
To validate the association between the newly identified susceptible loci and IgAN in a Korean population, we conducted a replication study with the two SNPs identified from the first stage in an independent sample of 310 IgAN cases and 438 healthy controls (Table 1). Of the two selected SNPs from the prior stage, one SNP showed significant associations with IgAN: rs2296136 in ANKRD16 (odds ratio [OR] = 1.40, 95% confidence interval [CI] 0.99–1.98, p = 0.05 in log-additive model, OR = 1.55, 95% CI = 1.06–2.27, p = 0.02 in dominant model, and OR = 0.70, 95% CI = 0.17–2.84, p = 0.62 in recessive model). We also analyzed the association between IgAN and the two selected SNPs in the entire study population (GWAS + replication study), and observed a significant association between IgAN and rs 2,296,136 in ANKRD16 (p = 0.0045 in log-additive model, p = 0.0027 in dominant model, and p = 0.76 in recessive model) (Table 3).
Associations between previously reported GWAS loci
We also performed an association analysis of IgAN with previously reported susceptible loci. Table 4 shows susceptible SNPs previously associated with IgAN and their references [6, 10, 11, 13,14,15,16]. Although we failed to demonstrate an association between IgAN and certain previously reported SNPs (rs6677604 in CFH, rs2523946 in HCG9, rs1883414 in HLA-DPB2, rs660895 in HLA-DRB1, rs2187668 in HLA-DQA1, rs2856717 in HLA-DQB1, rs2412971 in HORMAD2, rs11574637 in ITGAX, rs3803800 in TNFSF13, rs4227 in SOX15, rs252394 in MICD, and rs12537 in MTMR3), we found a modest association (p < 0.05) with other SNPs associated with IgAN in the HLA-DRB1 and HLA-DQB genes (Table 4).
Discussion
Here, we present the results of a two-stage GWAS involving 492 biopsy-confirmed IgAN cases and 893 healthy controls. Despite the small sample size, our study is valuable in that we aimed to overcome the limitations of earlier studies by using a customized DNA chip, which is predominantly composed of exon and promoter regions and contained not only well-established but also unknown SNPs. In addition, we selected patients with biopsy-confirmed IgAN to overcome the small sample size. With this approach, we identified new susceptible loci of IgAN in the Korean population. The primary contribution from our study are: 1) we designed a customized DNA chip containing 98,667 SNPs; 2) we genotyped two candidate SNPs selected in the discovery stage using a validation cohort; and 3) we identified one susceptible SNP; rs2296136 in ANKRD16.
Despite remarkable progress since IgAN was first described by Berger et al. in 1968 [17], its pathogenesis has not yet been clearly defined. Inter-individual variation of disease course, differences in incidence among different ethnicities, and familial aggregation of the disease have suggested a genetic predisposition for IgAN [5]. In the last two decades, there have been many candidate-gene association studies and linkage analyses for IgAN [6, 7]. However, those studies were underpowered, and no specific causative mutations for IgAN have been identified. GWASs have recently emerged as an alternative approach, allowing for the identification of susceptibility loci that were previously unrecognized [18].
The first GWAS of IgAN was performed in subjects of European ancestry by Feehally et al. [8]. This study provided evidence for an association between IgAN and genes at HLA loci, across HLA-B, DRB1, DQA, and DQB. In several subsequent GWASs, nearly 20 risk variants for IgAN were identified (CFHR1, CFHR3, HORMAD2, TNFSF13, DEFA,ITGAM-ITGAX, VAV3, and CARD9, among others) [9,10,11]. Those loci are associated with the complement system, mucosal IgA production, and innate and acquired immunity [2]. However, these previously reported SNPs are GWASs that were fixed and had less coverage of SNPs in the exon and promoter regions.
Recently, several large-scale GWASs on the population of East Asia have been reported. Yu et al. [11] conducted a GWAS to identify susceptibility loci for IgAN in Han Chinese and showed that IgAN is associated with SNPs of near genes involved in innate immunity. This study group also performed the largest GWAS of IgAN in Han Chinese and identified new susceptibility loci (rs7634389 in ST6GAL1, rs2074038 in ACCS, and rs2033562 in ODF1-KLF10) [16]. The results of these previous studies have helped clinicians understand the pathogenesis of IgAN. However, considering the genetic differences and prevalence between East Asian countries, it is necessary to conduct GWAS of IgAN in the Korean population.
In the present study, we used the Axiom™ Genome-Wide Human Assay and found two SNPs with suggestive evidence for association with IgAN in the first stage (p ≤ 5 × 10− 5). Among these, rs2296136 in ANKRD16 showed significant association with IgAN in the validation stage. ANKRD16 is located at 10p15.1 and encodes the ankyrin repeat domain 16. Its function is unclear because only a few studies have investigated ANKD16. One study showed that ANKRD16 is associated with subtype differences of breast cancer [19]. No study has reported an association between genetic variation in ANKRD16 and IgAN. We explored the effect of the variant on protein structure, function in missense SNPs and transcriptional activity in promoter SNPs. The probability of damage (probability > 0.8) for rs2296136 of ANKRD16 was validated by polyphen2. Further functional studies are needed to elucidate whether ANKRD16 can affect IgAN.
Performing validation study of GWAS results is important for extending the effect estimation and providing acceptable statistical evidence [20, 21]. Although our study had small sample size in the discovery stage, we also validated our results using an independent samples consisting of 310 biopsy-confirmed IgAN cases and 438 healthy controls.
Our study has some potential limitations. First, this GWAS was conducted in a relatively small patient population. Because of the small sample size, statistically significant SNPs of p < 1 × 10− 8 were not found. However, we found SNPs that were presumed to be related to IgAN and proceeded to validation. In the existing GWAS study, the most significant SNPs were mostly rare SNPs, and these significant SNPs were not significant when tested in other groups. Genetic polymorphic markers based on DNA in precision medicine are very important. Race, sex, and other factors affect the significance of these SNPs for any given disease. Therefore, we cannot say that the SNPs found in this study are statistically highly significant, but the SNPs reported through these studies may help to find additional markers. As described, to compensate for the sample size, we selected patients with biopsy-confirmed IgAN. Second, we focused only on SNPs with a minor allele frequency greater than 0.05 and so might have missed rarer variations associated with IgAN. Third, the patients included in this study were predominantly Korean, so the results should be generalized with caution. Finally, we did not assay gene expression in vivo or examine functional effects according to genetic variants, relying instead on in silico functional detection software. To improve these weakness, we are planning a follow-up study using expression quantitative trait loci (eQTL) analyses. Interestingly, however, both promoter and missense functional assay programs showed that the rs2296136 variant of ANKRD16 has important functional effects.
Conclusions
This study is the first to identify a significant association between IgAN and a customized GWAS chip containing mostly exons and promoter regions. Several susceptible genetic loci suggest that these significant SNPs may be useful for investigating the pathogenesis of IgAN. The SNPs identified in the present study clarify the genetic architecture of IgAN and point to new pathogenic pathways.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request.
Abbreviations
- CI:
-
Confidence interval
- eQTL:
-
Expression quantitative trait loci
- GWAS:
-
Genome-wide association study
- IgAN:
-
Immunoglobulin A nephropathy
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- OR:
-
Odds ratio
- Q-Q:
-
Quantile-quantile
- SNPs:
-
Single nucleotide polymorphisms
References
Wyatt RJ, Julian BA. IgA nephropathy. N Engl J Med. 2013;368:2402–14.
Magistroni R, D'Agati VD, Appel GB, Kiryluk K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015;88:974–89.
Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, Snyder HJ, Choi M, Hou P, Scolari F, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8:e1002765.
Kiryluk K, Novak J, Gharavi AG. Pathogenesis of immunoglobulin a nephropathy: recent insight from genetic studies. Annu Rev Med. 2013;64:339–56.
Kiryluk K, Julian BA, Wyatt RJ, Scolari F, Zhang H, Novak J, Gharavi AG. Genetic studies of IgA nephropathy: past, present, and future. Pediatr Nephrol. 2010;25:2257–68.
Wang W, Li G, Hong D, Zou Y, Fei D, Wang L. Replication of genome-wide association study identified seven susceptibility genes, affirming the effect of rs2856717 on renal function and poor outcome of IgA nephropathy. Nephrology (Carlton). 2017;22:811–7.
Xie J, Shapiro S, Gharavi A. Genetic studies of IgA nephropathy: what have we learned from genome-wide association studies. Contrib Nephrol. 2013;181:52–64.
Feehally J, Farrall M, Boland A, Gale DP, Gut I, Heath S, Kumar A, Peden JF, Maxwell PH, Morris DL, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. 2010;21:1791–7.
Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, Sanna-Cherchi S, Men CJ, Julian BA, Wyatt RJ, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43:321–7.
Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, Fasel D, Lata S, Prakash S, Shapiro S, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46:1187–96.
Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, Sun LD, Sim KS, Li Y, Foo JN, et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet. 2011;44:178–82.
Oh KH, Park SK, Park HC, Chin HJ, Chae DW, Choi KH, Han SH, Yoo TH, Lee K, Kim YS, et al. KNOW-CKD (KoreaN cohort study for outcome in patients with chronic kidney disease): design and methods. BMC Nephrol. 2014;15:80.
Zhu L, Zhai YL, Wang FM, Hou P, Lv JC, Xu DM, Shi SF, Liu LJ, Yu F, Zhao MH, et al. Variants in complement factor H and complement factor H-related protein genes, CFHR3 and CFHR1, affect complement activation in IgA nephropathy. J Am Soc Nephrol. 2015;26:1195–204.
Ferreira RC, Pan-Hammarstrom Q, Graham RR, Gateva V, Fontan G, Lee AT, Ortmann W, Urcelay E, Fernandez-Arquero M, Nunez C, et al. Association of IFIH1 and other autoimmunity risk alleles with selective IgA deficiency. Nat Genet. 2010;42:777–80.
Yang C, Jie W, Yanlong Y, Xuefeng G, Aihua T, Yong G, Zheng L, Youjie Z, Haiying Z, Xue Q, et al. Genome-wide association study identifies TNFSF13 as a susceptibility gene for IgA in a south Chinese population in smokers. Immunogenetics. 2012;64:747–53.
Li M, Foo JN, Wang JQ, Low HQ, Tang XQ, Toh KY, Yin PR, Khor CC, Goh YF, Irwan ID, et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat Commun. 2015;6:7270.
Berger J, Hinglais N. Intercapillary deposits of IgA-IgG. J Urol Nephrol (Paris). 1968;74:694–5.
Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J Clin Invest. 2014;124:2325–32.
O'Brien KM, Cole SR, Engel LS, Bensen JT, Poole C, Herring AH, Millikan RC. Breast cancer subtypes and previously established genetic risk factors: a bayesian approach. Cancer Epidemiol Biomark Prev. 2014;23:84–97.
Konig IR. Validation in genetic association studies. Brief Bioinform. 2011;12:253–8.
Kraft P, Zeggini E, Ioannidis JP. Replication in genome-wide association studies. Stat Sci. 2009;24:561–73.
Acknowledgements
The authors acknowledge all of the patients who participated in the KNOW-CKD and the investigators who took part in data collection.
The authors thank Tae Won Lee and Chun Gyoo Ihm for their fruitful scientific advice.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (NRF-2017R1A2B4011623).
Funding
No funding was received.
Author information
Authors and Affiliations
Consortia
Contributions
YHK and SHL designed the study. KHJ, JSK, and SKK performed the experiments and analyzed data. KHJ, JSK, YHK, and SHL wrote the paper. YHL, YGK, JYM, SWK, and THK participate in discussion. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the institutional review board of all centers, and the approval number was 2012–01-130 obtained from Kyung Hee University Hospital at Gangdong. Written informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors have no competing interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jeong, K.H., Kim, J.S., Lee, Y.H. et al. Genome-wide association study identifies new susceptible loci of IgA nephropathy in Koreans. BMC Med Genomics 12, 122 (2019). https://doi.org/10.1186/s12920-019-0568-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-019-0568-6