
Abstract
Background
Multifunctional transcription factor (TF) gene EWS/EWSR1 is involved in various cellular processes such as transcription regulation, noncoding RNA regulation, splicing regulation, genotoxic stress response, and cancer generation. Role of a TF gene can be effectively studied by measuring genome-wide gene expression, i.e., transcriptome, in an animal model of Ews/Ewsr1 knockout (KO). However, when a TF gene has complex multi-functions, conventional approaches such as differentially expressed genes (DEGs) analysis are not successful to characterize the role of the EWS gene. In this regard, network-based analyses that consider associations among genes are the most promising approach.
Methods
Networks are constructed and used to show associations among biological entities at various levels, thus different networks represent association at different levels. Taken together, in this paper, we report contributions on both computational and biological sides.
Results
Contribution on the computational side is to develop a novel computational framework that combines miRNA-gene network and protein-protein interaction network information to characterize the multifunctional role of EWS gene. On the biological side, we report that EWS regulates G-protein, Gnai1, in the spinal cord of Ews/Ewsr1 KO mice using the two biological network integrated analysis method. Neighbor proteins of Gnai1, G-protein complex subunits Gnb1, Gnb2 and Gnb4 were also down-regulated at their gene expression level. Interestingly, up-regulated genes, such as Rgs1 and Rgs19, are linked to the inhibition of Gnai1 activities. We further verified the altered expression of Gnai1 by qRT-PCR in Ews/Ewsr1 KO mice.
Conclusions
Our integrated analysis of miRNA-transcriptome network and PPI network combined with qRT-PCR verifies that Gnai1 function is impaired in the spinal cord of Ews/Ewsr1 KO mice.
Similar content being viewed by others


Background
Ewing sarcoma is the second most common bone and soft tissue tumor that predominantly afflicts children and adolescents [1–3]. Understanding biological mechanisms underlying this tumor is critical to the identification of new cancer therapy targets. The Ewing sarcoma gene (EWS)/EWS RNA-Binding Protein 1 (EWSR1), a transcription factor, encodes an RNA binding protein whose specific functional targets are still largely unknown [4]. In previous studies, fusion genes such as, EWS-FLI-1, EWSR1-WT1, EWSR1-KLF17, EWSR1-ATF1, and EWSR1-CREB3L1, are known to be produced by rearrangement of the EWSR1 gene with different gene fusion partners and these fusion genes have functions related to a variety of soft tissue tumors [5–9]. To characterize functions of EWS, we used RNA-seq gene expression data and miRNA expression data measured by using the spinal cord samples of Ews/Ewsr1 knock-out (KO) mouse and wild type.
Motivation
Multi-function genes interact with a number of coding and non-coding genes and perform a variety of functions depending on cell conditions and tissue types. Multi-function gene EWSR1 is known to regulate Drosha and microRNAs that inhibits RNA splicing [10, 11]. However, it is still unknown which genes are regulated by and which biological functions are related to EWSR1. To characterize functions of EWSR1, we used a well-known differentially expressed gene (DEG) set analysis. We performed functional analysis of top 200 up-regulated DEGs and top 200 down-regulated DEGs (2 % of the whole genes) using gene ontology (GO) and KEGG pathway. From the GO analysis, we found 322 genes of 400 top DEGs were involved in 44 GO terms in the GOTERM_BP_FAT category which is the summarized version of Biological Processes in the Gene Ontology (Additional file 1A). Top three GO terms with the largest number of genes were ion transport, immune response, and homeostatic process. It is not clear how these three biological processes are related to EWS. In addition, we tried molecular function GO terms, which did not produce coherent biological functions related to EWS. From the KEGG pathway result, 93 of 400 genes hit 140 pathways. Only two pathways had more than 10 genes: metabolic pathway and cell adhesion molecules. Most of the pathways were not significant. Overall, GO and KEGG pathway analysis using DEGs did not produce meaningful clues on the role of EWS.
For the analysis of miRNA expression data, it is not clear how to perform an integrated analysis of gene expression data and miRNA expression data. In addition, a multifunction gene can play roles at various levels such as transcription, gene regulation, translation and protein activity level. To address this computational challenge, we developed a novel computational framework for the characterization of EWS multifunctional gene using gene expression data and miRNA expression data measured under a knockout condition of the multifunctional gene. The framework utilized microRNA-target gene network and Protein-Protein interaction (PPI) network and incorporates the two networks in a workflow. The workflow of the framework can be viewed as an effort to model the role of EWS at various levels, DEG analysis at the transcription level, the microRNA-target gene network analysis at the gene regulation level, and PPI network analysis at the translation and protein activity level.
Methods
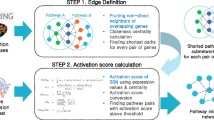
We developed a three-step pipeline for the integrated analysis of omics data using mRNA-microRNA network and protein-protein interaction network. We describe the workflow and computational methods used in each step in this section. Figure 1 illustrates the workflow of the proposed omics data analysis pipeline. In “Results” section, we discuss output from each step in detail.

Illustration of the workflow of the pipeline. Transcription factor (TF) gene has multiple functions to regulate transcription. Generated mRNAs are regulated by microRNA and translated proteins have functions with interacted proteins and molecules. RNA sequencing data and microRNA (miRNA) microarray data are generated from spinal cord extraction in Ews/Ewsr1 knockout and wild type mice. SAM (Significance Analysis of Microarrays) is used for selection of significantly expressed miRNA from miRNA microarrays. TargetScan and miRDB were used to predict target genes of miRNAs. From RNA sequencing data, gene expression values are mapped to the reference genome data using Tophat. Then negative correlated differentially expressed genes (DEGs) are selected. Significantly expressed microRNA target genes have many interacting proteins. Specific target gene interactional neighbor proteins are searched in the STRING DB. PPI network analyzed with gene expression value. Analysis results of miRNA-mRNA network and PPI network are integrated by analyzing correlation in expression levels. Regulated genes further are analyzed and visualized with DAVID, KEGG and Cytoscape
Step 1. MicroRNA-target gene regulation network analysis
-
Input: gene expression data, miRNA expression data
-
Output: differentially expressed miRNAs and their target genes
To investigate roles of EWS, we analyzed the translational regulatory network. The microRNA-target gene integrated network analysis was performed following the strategy in MMIA [12].
Selection significantly expressed microRNAs
We selected significantly up- or down-regulated microRNAs in the Ews/Ewsr1 KO condition compared to the wild type condition. To select significantly differentially expressed miRNAs from microarray data, we used the SAM (significance analysis of microarrays) tool package [13] (More information in the detailed method section).
Prediction of microRNAs target genes
After selecting significantly expressed microRNAs, we predicted regulatory target genes of the selected differentially expressed microRNA by TargetScan [14] and miRDB [15, 16].
Reselection target genes by correlation
We further investigated miRNA and gene target relationship by measuring negative correlation in expression levels between miRNAs and genes targeted by miRNAs since up-regulated microRNA inhibits translation of mRNA.
Step 2. Pathway analysis of DEGs from MMIA analysis and validation
-
Input: DEGs selected in Step 1
-
Output: important pathways related to EWS and key genes in the pathways
Differentially Expressed Gene (DEG) analysis
Differentially expressed genes (DEGs) analysis of NGS RNA-seq was performed in the following steps. First, adaptor sequences of reads in raw data were trimmed. The Ensembl mouse reference genome sequence was downloaded for mapping short reads. Bowtie [17] was used to build an index of the reference genome sequence for alignment. Trimmed reads were then mapped to the reference genome sequence using Tophat2 [18]. Finally, Cufflinks was used to calculate gene expression levels. We compared gene expression values and selected DEGs by using Cuffdiff in the Cufflinks package [19].
Integrated analysis of miRNA and mRNA expression data
15 differentially expressed miRNAs were found to target 4342 genes based on TargetScan and miRDB. To further screen target genes, we integrated miRNAs target information and mRNA-seq based gene expression levels. The negative correlation analysis reduced the number of targets to 1338 genes. The negative correlation analysis is based on the techniques in [20, 21]. The rationale for the negative correlation analysis is that if a miRNA targets a gene the expression levels of the miRNA and the gene should have negative correlation due to the regulatory effect of miRNA on the target gene. These DEGs were then analyzed by GSEA (Gene Set Enrichment Analysis) using DAVID (The Database for Annotation, Visualization and Integrated Discovery) [22].
Pathway analysis
To characterize functions of selected target DEGs by negative correlation in the spinal cord of Ews/Ewsr1 KO mice, we performed biological pathway analysis using the KEGG mapper [23]. KEGG mapper highlighted DEGs with colors: up-regulated DEGs as red, down-regulated DEGs as blue, and other DEGs as light green. In addition, we performed additional pathway interpretation based on gene ontology by using ClueGO [24], a Cytoscape [25] plug-in, that analyzes biological pathway interpretation with KEGG ontology (2014 latest version) to integrate Gene Ontology (GO) terms and KEGG/BioCarta pathways to generate a functionally organized GO/pathway term network.
Verification of Gnai1 expression by Quantitative real-time PCR (qRT-PCR)
To verify whether the expression of target genes is correlated with the analysis, we performed qRT-PCR using RNA isolated from the spinal cords of Ews/Ewsr1 WT and KO mice.
Step 3. Protein-protein interaction network analysis
-
Input: Key genes identified in Step 2
-
Output: G protein complex genes and regulators
After selecting the key gene in Step 2, we investigated the biological functions of the genes by extending gene sets with neighboring genes of the key gene.
Selection significantly expressed gene
From gene set analysis (GSA) and pathway analysis (see the detailed methods section), we selected specific genes.
Search for proteins that interact with the selected gene
Protein-protein interaction (PPI) analysis of genes neighboring the key gene was performed by using STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) [26], the most widely used database of known and predicted protein interactions.
Analysis of biological functions
Relationship between the key gene and neighbor genes was investigated by performing the literature search. When we considered the relationship among genes, we also considered the regulatory roles of genes, i.e., activators or repressors, if applicable. For the regulatory relationship, we considered gene expression change information.
Results
Analysis of multifunctional EWS by using the network-based workflow
In this section, we present the result from each computational step of the workflow (Fig. 1).
Step 1. Translational regulatory network analysis: MicroRNA-mRNA network
Selection of differentially expressed miRNAs
We selected 18 significantly expressed miRNAs from the total 1193 mouse miRNAs by SAM tool. 15 miRNAs expression level were significantly up-regulated, and 3 miRNAs were down-regulated in the Ews/Ewsr1 KO mice against WT mice (Additional file 2). In the order of the significance score by SAM, 15 up-regulated miRNAs are mmu-miR-127, mmu-miR-410, mmu-miR-433, mmu-miR-138, mmu-miR-181c, mmu-miR-382, mmu-miR-19b, mmu-miR-381, mmu-miR-666-3p, mmu-miR-376a, mmu-miR-873, mmu-miR-181a, mmu-miR-383, mmu-miR-181b, and mmu-miR-99b. Down-regulated 3 miRNAs were mmu-miR-1224, mmu-miR-9-3p, and mmu-miR-26a in the order of the significance score by SAM. Analysis of potential biological functions of these miRNAs was performed by using genes targeted by the miRNAs (see the DEG analysis from RNA-seq data result section).
Prediction of target mRNA regulated by selected miRNA
To perform the integrated analysis of miRNA and their target genes, we need to predict targets of miRNAs. Predicted target genes of miRNAs were collected by using TargetScan and miRDB. 5,779 and 5,448 genes were predicted by TargetScan and miRDB, respectively. 1,927 genes were targeted by multiple miRNAs in the prediction result of TargetScan, and 2,371 genes were multiply targeted according to miRDB. After discarding repeatedly predicted genes, a total of 4,342 genes were predicted as targets of 15 differentially expressed miRNAs. Only 36 % (1,587 genes) of predicted target genes were predicted by both TargetScan and miRDB. In other words, the genes targeted by each miRNAs of prediction results by TargetScan and miRDB do not agree much (Additional file 3). 4,342 target genes predicted by both TargetScan and miRDB were further analyzed by performing a negative correlation analysis to sort out potentially true miRNA-gene relationships (see the next section).
Negative correlation analysis of DEGs with DE microRNA
Predicted target genes were further screened by considering negative correlations in expression levels between miRNA and each of its target genes. The rationale for the negative correlation analysis is that miRNA degrades its target genes, thus a higher expression level of miRNA should result in a lower expression level of its target. We applied the same technique used in [14, 15]. Negatively correlated miRNA-mRNA interaction network of miRNAs and their target DEGs were visualized by using Cytoscape (Fig. 2). In Fig. 2, significantly up-regulated 15 miRNAs are in red color, and negative correlated target DEGs are in blue color. Color intensity denoted the level of gene expression. As a result of the correlation analysis, 4,342 genes were reduced to 860 genes. Among the 860 DEGs, 339 target genes were targeted by multiple miRNAs.

Network of microRNAs and mRNAs. Up-regulated miRNAs (Red nodes) are selected by SAM. Target genes (mRNAs, blue nodes) of selected miRNAs are predicted by TargetScan (left) and miRDB (right). Down-regulated genes targeted by up-regulated miRNA are selected from each predicted results. miRNA-mRNA interaction network is drawn by Cytoscape. Color intensity denotes the level of gene expression
Step 2. Pathway analysis of DEGs from MMIA analysis and validation
KEGG pathway analysis of DEGs gene set targeted by miRNA
We mapped the 860 negatively correlated DEGs to the KEGG pathway using the KEGG mapper. 201 pathways were hit by the negatively correlated DEGs. We selected 13 pathways with eight or more gene hits. Metabolic pathways, calcium signaling pathway, PI3K-Akt signaling pathway, axon guidance, pathways in cancer, MAPK signaling pathway, tight junction, dilated cardiomyopathy, circadian entrainment, proteoglycans in cancer, regulation of actin cytoskeleton, cholinergic synapse and focal adhesion pathways were selected. Analysis of KEGG pathways of DEGs were highlighted in colors chosen by KEGG mapper. Blue color genes were down-regulated genes, and red color genes were up-regulated genes in the pathways of Ews/Ewsr1 KO mice (Additional file 4). Color intensity denoted the level of gene expression.
Gene ontology based network analysis
Networks of negatively correlated target DEGs in terms of KEGG ontology were generated using ClueGO (Fig. 3). “Cholinergic synapse pathway” term was highly clustered by down-regulated DEGs belonging pathways. ECM-receptor interaction pathway, focal adhesion pathway, tight junction pathway, and action cytoskeleton regulation pathway were mostly correlated with selected down-regulated DEGs. Gnai1, which is most significantly down-regulated in the cholinergic synapse pathway, was selected for further investigation. More discussion on biological functions of these pathways is presented in the Conclusion section.

Venn diagram generated by ClueGO. ClueGO analyzes KEGG ontology of selected down-regulated genes which are targeted by up-regulated miRNA. Cholinergic synapse pathway is showed highly clustered by down-regulated gene pathways
qRT-PCR of Gnai1
qRT-PCR was performed to confirm the difference of Gnai1 expression in the spinal cords of Ews/Ewsr1 WT and KO mice. Average gene expression levels of Gnai1 in Ews/Ewsr1 KO mice were significantly lower than those in Ews/Ewsr1 WT mice. This data validated that Gnai1 expression level was down regulated in Ews/Ewsr1 KO mice (Fig. 4).

Verification of altered Gnai1 expression in Ews/Ewsr1 WT and KO mice. The gene expression level of Gnai1 was significantly lower in the spinal cords of Ews/Ewsr1 KO mice (n = 6) compared to EWS WT mice (n = 6). The bar graph represents average ± standard error mean (SEM). **, Significantly different at p < 0.01 by Student T-test
Step 3. Protein-protein interactions network analysis
We selected Gnai1 that is down-regulated in cholinergic synapse pathways and action cytoskeleton regulation pathway. To investigate the effect of down-regulation of Gnai1, we used the STRING protein-protein interaction network DB. In the PPI network, genes neighboring Gnai1 were further investigated for their biological functions. Looking at gene expression values, we were able to confirm the relationship between G-protein genes and RGS genes. Genes neighboring Gnai1 were selected by using STRING (Fig. 5). Top 20 interacted genes are shown in Table 1. Gnai1 and G-protein related genes, such as Gnb1, Gnb2 and Gnb4, were down-regulated at their gene expression level (Fig. 6). In contrast, Rgs1 and Rgs19, regulators of G-protein signaling genes that are associated with the inhibition of Gnai1 function, were up-regulated (Fig. 6).

PPI network of Gnai1 from the STRING DB

G-proteins and RGS (regulator of G-protein) expression level and log2 fold change value in Ews/Ewsr1 wild type and knock-out
Discussion
Potential interaction map of EWS, RGS, and G-protein complex genes
A growing body of evidence shows multifunctional roles of the EWS/EWSR1 fusion oncoproteins [5, 7–9]. However, the role of wild-type (WT) EWS/EWSR1 is not fully understood yet. EWS/EWSR1 deficiency contributes to the failure of precursor B lymphocyte development and leads to the premature cellular senescence in mouse embryonic fibroblasts (MEFs) [27, 28]. It seems likely that the WT EWS/EWSR1 protein exhibits many different cellular functions in a cell-type specific manner. In the spinal cord of Ews/Ewsr1 KO mice, microRNAs, such as mmu-miR-381 and mmu-miR-181a/b/c were up-regulated. These microRNAs suppressed expression of Gnai1 (Gi Protein Alpha subunit). Concurrently, RGS (Regulator of G-protein Signaling) genes, Rgs1 and Rgs19, were up-regulated, which repressed Gnai1 activity. In addition, G Protein Beta subunit genes, Gnb1, Gnb2 and Gnb4 were down-regulated. Thus in the Ews/Ewsr1 KO condition, G protein complex was not formed (Fig. 7).

Roles of G proteins and its regulatory mechanisms by miRNAs in the spinal cord of Ews/Ewsr1 KO mouse. Direction of arrow means with a change of gene expression level in Ews/Ewsr1 KO mice. Upper arrows are up-regulated gene expression level, and bottom arrows are the opposite
Since Gnai1 was down-regulated, it is proposed that Gnai1 may be unable to inhibit downstream adenylate cyclase genes, such as Adcy9 and Adcy4, in cholinergic synapse pathway. Adenylate cyclase catalyzes the conversion of ATP to cAMP, and the cAMP regulates cAMP-proteins, transcription factors, and cAMP-dependent kinases. Adenylate cyclase is an enzyme with key regulatory roles, and Adenylate cyclase regulator Gnai1 has important roles in cholinergic synapse.
Our study presents for the first time that Ews/Ewsr1 deficiency modulates microRNA processing in the spinal cord. Notably, increased levels of mmu-miR-381 and mmu-miR-181a/b/c were directly associated with the down regulation of G protein complex in the spinal cord of Ews/Ewsr1 KO mice. We have previously shown that Ews/Ewsr1 deficiency leads to abnormal microRNA processing and skin development via Drosha-dependent pathway [10]. Furthermore, we found that Ews/Ewsr1 deficiency reduces the expression of Uvrag (UV radiation resistance associated) gene at the post-transcription level via mmu-miR-125a and mmu-miR-351 [29]. Interestingly, the reduction of Uvrag by mmu-miR-125a and mmu-miR-351 impaired autophagy function in Ewsr1 knockout (KO) MEFs and KO mice. Considering that G protein-coupled signaling transduction pathway is very complex, the Gnai1-dependent cellular function and mechanism in in vitro and in vivo models of EWSR1 deficiency remains to be determined in future studies.
Conclusion
We developed a computational framework for the analysis of the multifunction TF EWS gene and showed that EWS has a significant role in the regulation of G protein complex. Since a multifunction TF gene has a complicated biological functions at various levels, such as transcription, gene regulation, and protein levels, powerful analysis tools are needed. Our method utilized miRNA-target gene network and protein-protein interaction network and combined multiple tools in a single computational framework.
We analyzed the miRNAs and mRNA data in the spinal cord of Ews/Ewsr1 KO mice, and selected all significantly differentially expressed miRNAs and negative correlated DEGs. We constructed an interaction network with selected miRNAs and mRNAs and analyzed the GSEA and related pathways. From the result of pathway analysis, we identified significantly down-regulated Gnai1 gene in the cholinergic synapse pathway that is highly clustered by down-regulated DEGs belonging pathways. Gnai1 was verified by qRT-PCR, and analyzed about PPI sub-networks. Gnai1 was suppressed by mmu-miR-381 and mmu-miR-181a/b/c, and inhibited by Rgs1 and Rgs19 in the spinal cord of Ews/Ewsr1 KO mice. As a future work, we plan to develop a software package for the analysis of multifunction TF genes.
Material & detailed methods
NGS data
RNA sequencing data and microRNA microarray data those were generated from the spinal cord tissue samples of Ews/Ewsr1 WT and KO mice [10].
Differentially expressed miRNA analysis
Differentially expressed miRNAs were selected from miRNA microarray data by using the samr (SAM: Significance Analysis of Microarrays, version: 2.0) package in Bioconductor. We used “two-class unpaired” option with 1000 permutations. SAM generated an interactive plot of the observed vs. expected (based on the permuted data) d-values. The user can dynamically change thresholds for significance to set the value of the tuning parameter delta. We set the delta to 2 to reduce the numbers of selected significant miRNAs.
MicroRNA target Gene prediction
We collected target genes of differentially expressed miRNAs using TargetScan and miRDB. TargetScan predicts biological targets of selected miRNAs by searching for the presence of conserved 8mer and 7mer sites that match the seed region of each miRNA. miRDB is a database of predicted miRNA targets in animals. MicroRNA targets in miRDB were predicted by using SVM (support vector machine) based prediction program. Only 22 % of predicted target genes by TargetScan and miRDB agreed. Since we were unable to decide which predicted gene are correct and we used all predicted target genes.
Reference genome sequence for alignment
We downloaded and used Ensembl reference genome sequence (Mus_musculus.GRCm38.70) for reads mapping [30].
GTF (General Transfer Format) file for gene annotation
After the alignment, we calculated the FPKM (fragment per kb exon model) values of each gene by Cufflinks with Ensembl gene model (Mus_musculus.GRCm38.70) [31].
Preprocessing of RNA-sequence data for DEG analysis
Before mapping reads, we clipped two adaptor sequences of paired-end RNA-seq data. For trimming, we allowed 2 mismatch of adaptor sequences to short reads. After the trimming process, we discarded reads of 18 bp or shorter.
Used trimming processing adaptor sequences show the next lines.
READ1 adaptor sequence: GATCGGAAGAGCACACGTCTGAACTCCAGTCAC
READ2 adaptor sequence: AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTATCATT
DEG (differently expressed gene) analysis from RNA-seq NGS data
Paired-end total RNA-sequencing raw data were generated by Illumina HiSeq 2000. Each of the numbers of reads in raw data of wild type and Ews Knockout 3-week-old mice spinal cord samples show Table 2. After adaptor sequence trimming process for discarding of low quality sequence, the number of trimmed reads for each samples show Table 2. These amount of reads is sufficient for DEG analysis. After reference genome indexing, trimmed short reads were mapped to the reference genome by Tophat. The ratios of mapped reads for each samples were 81.72 and 81.5 %. The mapping ratios were higher than 80 % for all samples and variations in the mapping ratio across the samples were very small. Thus we believe that results of analysis for RNA sequencing experiment and short read processing were satisfactory. We quantified the expression level of each gene using Cufflinks based on the gene information from Ensembl.
Quantitative real-time PCR
Total RNA was extracted from the spinal cord of Ews/Ewsr1 WT and KO mice by TRIzol reagent (MRC, Cincinnati, OH, USA) as previously described [10]. RNA was measured in a spectrophotometer at 260-nm absorbance. RNA analysis was conducted as follows. Fifty nanograms of RNA were used as a template for qRT-PCR amplification, using SYBR Green Real-time PCR Master Mix (Toyobo, Osaka, Japan). Primers were standardized in the linear range of cycle before the onset of the plateau. Mouse GAPDH was used as an internal control. Two-step PCR thermal cycling for DNA amplification and real-time data acquisition were performed with an ABI StepOnePlus Real-Time PCR System using the following cycle conditions: 95 °C for 1 min × 1 cycle, and 95 °C for 15 s, followed by 60 °C for 1 min × 40 cycles. Fluorescence data were analyzed by the ABI StepOnePlus software and expressed as, Ct, the number of cycles needed to generate a fluorescent signal above a predefined threshold. The ABI StepOnePlus software set baseline and threshold values.
Abbreviations
DAVID, the database for annotation, visualization and integrated discovery; DEG, differentially expressed gene; DNA, deoxyribonucleic acid; EWS, Ewing’s Sarcoma; EWSR1, EWS RNA-binding protein 1; FPKM, fragments per kilobase of exon per million fragments mapped; Gnai1, Gi protein alpha subunit; GO, gene ontology; GSEA, gene set enrichment analysis; KEGG, Kyoto encyclopedia of genes and genomes; KO, knock-out; MMIA, microRNA and mRNA integrated analysis; PPI, protein-protein interaction; qRT-PCR, quantitative real-time PCR; RGS, regulator of G-protein signaling; RNA, ribonucleic acid; RNA-seq, whole transcriptome sequencing; STRING, Search Tool for the Retrieval of Interacting Genes/Proteins; TF, Transcription factor
References
Meltzer PS. Is Ewing’s sarcoma a stem cell tumor? Cell Stem Cell. 2007;1(1):13–5.
Barker LM, Pendergrass TW, Sanders JE, Hawkins DS. Survival after recurrence of Ewing’s sarcoma family of tumors. J Clin Oncol. 2005;23:4354–62.
Miser JS, Krailo MD, Tarbell NJ, et al. Treatment of metastatic Ewing’s sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide–a Children’s Cancer Group and Pediatric Oncology Group study. J Clin Oncol. 2004;22:2873–6.
Bertolotti A, Bell B, Tora L. The N-terminal domain of human TAFII68 displays transactivation and oncogenic properties. Oncogene. 1999;18(56):8000–10.
May WA, Lessnick SL, Braun BS, Klemsz M, Lewis BC, Lunsford LB, Hromas R, Denny CT. The Ewing's sarcoma EWS/FLI-1 fusion gene encodes a more potent transcriptional activator and is a more powerful transforming gene than FLI-1. Mol Cell Biol. 1993;13(12):7393–8.
Fisher C. The diversity of soft tissue tumours with EWSR1 gene rearrangements: a review. Histopathology. 2014;64(1):134–50.
Huang SC, Chen HW, Zhang L, Sung YS, Agaram NP, Davis M, Edelman M, Fletcher CD, Antonescu CR. Novel FUS‐KLF17 and EWSR1‐KLF17 fusions in myoepithelial tumors. Genes Chromosomes Cancer. 2015;54(5):267–75.
Rossi S, Szuhai K, Ijszenga M, Tanke HJ, Zanatta L, Sciot R, Fletcher CD, Dei Tos AP, Hogendoorn PC. EWSR1-CREB1 and EWSR1-ATF1 fusion genes in angiomatoid fibrous histiocytoma. Clin Cancer Res. 2007;13(24):7322–8.
Lau PP, Lui PC, Lau GT, Yau DT, Cheung ET, Chan JK. EWSR1-CREB3L1 gene fusion: a novel alternative molecular aberration of low-grade fibromyxoid sarcoma. Am J Surg Pathol. 2013;37(5):734–8.
Kim KY, Hwang YJ, Jung MK, Choe J, Kim Y, Kim S, Lee CJ, Ahn H, Lee J, Kowall NW, Kim YK, Kim JI, Lee SB, Ryu H. A multifunctional protein EWS regulates the expression of Drosha and microRNAs. Cell Death Differ. 2014;21(1):136–45.
Chansky HA, Hu M, Hickstein DD, Yang L. Oncogenic TLS/ERG and EWS/Fli-1 fusion proteins inhibit RNA splicing mediated by YB-1 protein. Cancer Res. 2001;61(9):3586–90.
Nam S, Li M, Choi K, Balch C, Kim S, Nephew KP. MicroRNA and mRNA integrated analysis (MMIA): a web tool for examining biological functions of microRNA expression. Nucleic Acids Res. 2009, gkp294
Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98(9):5116–21.
Benjamin PL, Christopher BB, David PB. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell. 2005;120:15–20.
Xiaowei W, Issam MEN. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24(3):325–32.
Xiaowei W. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14(6):1012–7.
Ben L, Cole T, Mihai P, Steven LS. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2011;14:R36.
Trapnell C, Williams BA, Pertea G, Mortazavi AM, Kwan G, van Baren MJ, Salzberg SL, Wold B, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5.
Xin F, Li M, Balch C, Thomson M, Fan M, Liu Y, Hammond SM, Kim S, Nephew KP. Computational Analysis of MicroRNA Profiles and Their Target Genes Suggests Significant Involvement in Breast Cancer Antiestrogen Resistance. Bioinformatics. 2009;25(4):430–4.
Marbach D, Costello JC, Kuffner R, Vega NM, Prill RJ, Camacho DM, Allison KR, Kellis M, Collins JJ, Stolovitzky G. Wisdom of crowds for robust gene network inference. Nat Methods. 2012;9(8):796–804.
Dennis Jr G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4(5):3.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Gabriela B, Bernhard M, Hubert H, Pornpimol C, Marie T, Amos K, Wolf-Herman F, Franck P, Zlatko T. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25(8):1091–3.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Snel B, Lehmann G, Bork P, Huynen MA. STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 2000;28(18):3442–4.
Li H, Watford W, Li C, Parmelee A, Bryant MA, Deng C, et al. Ewing sarcoma gene EWS is essential for meiosis and B lymphocyte development. J Clin Invest. 2007;117:1314–1323.
Cho J, Shen H, Yu H, Li H, Cheng T, Lee SB, et al. Ewing sarcoma gene Ews regulates hematopoietic stem cell senescence. Blood. 2011; 117(4):1156–1166.
Kim YH, Kim KY, Hwang YJ, Kowall NW, Lee SB, Lee J and Ryu H. Uvrag targeting by MiR-125a and MiR-351 modulates autophagy associated with EWS deficiency. Autophagy. 2015; 11(5):796–811.
Available: ftp://ftp.ensembl.org/pub/release-70/fasta/mus_musculus/dna/. Accessed 1 May 2015.
Available: ftp://ftp.ensembl.org/pub/release-70/gtf/mus_musculus/. Accessed 1 May 2015.
Declaration
Publication of this article has been funded by the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Ministry of Science, ICT & Future Planning (2012M3A9D1054622); by the Bio & Medical Technology Development Program of the NRF funded by the Korean government, MSIP (NRF-2014M3C9A3063541); by the Next-Generation Information Computing Development Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (No.NRF-2012M3C4A7033341). This study was supported by NIH Grant (NS067283 to H.R.). This study was and Brain Science Flagship Grant (2E25480 to H.R.) from Korea Institute of Science and Technology.
This article has been published as part of BMC Medical Genomics Volume 9 Supplement 1, 2016. Selected articles from the 5th Translational Bioinformatics Conference (TBC 2015): medical genomics. The full contents of the supplement are available online https://bmcmedgenomics.biomedcentral.com/articles/supplements/volume-9-supplement-1
Availability of data and materials
All datasets on which the conclusions of the manuscript are presented in the main paper and additional supplementary files.
Authors’ contributions
SK and HR designed and supervised the research project and edited the paper. CL developed the method, performed data analysis and wrote the manuscript. HA helped data analysis and advised and assisted the manuscript. SBL, J-YS, W-YP, J-IK and JL supported to generate RNA-seq data and performed biological experiment. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The animal study using Ewsr1 WT and KO mice was approved by IACUC and animal experiments were conducted in accordance with the Guide for Institutional Animal Care and Use Committee of Seoul National University.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Top 400 DEGs analysis result. A) GO term result of DAVID analysis. B) KEGG pathway result list. (XLSX 19 kb)
Additional file 2: Figure S1.
Graphic plotting of miRNA microarray analysis by SAM. Red dots are significantly up-regulated miRNAs and green dots are down-regulated. In the table of SAM result, columns are score, numerator, denominator, fold change and q-value. (DOCX 34 kb)
Additional file 3: Table S2.
The number of genes targeted by each miRNAs by using TargetScan and miRDB. Prediction results by TargetScan and miRDB do not agree much. Union of target genes were further analyzed by performing. (DOCX 15 kb)
Additional file 4: Figure S2.
The cholinergic synapse pathway related with significantly down-regulated genes by ClueGO. Selected DEGs are highlighted in colors chosen by KEGG mapper. Blue genes are down-regulated genes, and red genes are up-regulated genes in Ews/Ewsr1 KO mice compared to WT mice. Green color genes are not changed. (DOCX 53 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lee, CJ., Ahn, H., Lee, S.B. et al. Integrated analysis of omics data using microRNA-target mRNA network and PPI network reveals regulation of Gnai1 function in the spinal cord of Ews/Ewsr1 KO mice. BMC Med Genomics 9 (Suppl 1), 33 (2016). https://doi.org/10.1186/s12920-016-0195-4
Published:
DOI: https://doi.org/10.1186/s12920-016-0195-4