
Abstract
Background
Whereas genome-wide association study (GWAS) has proven to be an important tool for discovery of variants influencing many human diseases and traits, unfortunately its performance has not been much of all-around success for some complex conditions, for example, hypertension. Because some of the existing effective pharmacotherapeutic agents act by targeting known biological pathways, pathway-based analytical approaches could lead to more success in discovery of disease-associated variants. The objective of the present study was to identify functional variants associated with blood pressure in the aldosterone-regulated sodium reabsorption pathway using the simulated and real blood pressure phenotypes provided for Genetic Analysis Workshop 19.
Methods
The present analysis included 1942 samples with exome sequencing data and for whom blood pressure phenotypes were available. Because only odd-numbered autosomes were available, we restricted analysis to 127 quality-controlled single-nucleotide polymorphisms from the aldosterone-regulated sodium reabsorption pathway. We performed pathway-based association analysis using appropriate regression models for single variant, haplotype and epistasis association analyses. To account for multiple comparisons, statistical significance was empirically derived by permutation procedure and Bonferroni correction.
Results
The topmost pathway-based association signals were observed in PRKCA gene for diastolic blood pressure (DBP), systolic blood pressure (SBP), and mean arterial pressure (MAP) in both real and simulated data. The associations remained significant (P <0.05) after multiple testing corrections for the number of genes. Similarly, the pathway-based 2-locus epistasis analysis indicated significant interactions between INSR and PRKCG for SBP and MAP; INS and PIK3R2 for DBP; PIK3CD and ATP1B2 for hypertension in the real data set. We also observed significant within-gene interactions in INSR for SBP, DBP, and hypertension in the simulated data set.
Conclusion
The findings from this study show that pathway-based analytical approach targeting known biological pathways can be useful in identification of disease-associated variants that are otherwise undetectable by GWAS. The approach takes advantage of the assumption of nonindependence of variants within and across pathway genes which leads to reduced penalty of multiple testing and thus less-stringent statistical significance threshold.
Similar content being viewed by others


Background
Genome-wide association study (GWAS) has proved to be a useful tool in the discovery of genetic variants associated with many complex diseases and traits [1]. Unfortunately, the level of success in variants discovery by GWAS for some complex human diseases, such as elevated blood pressure or hypertension, has been very low. In fact, variants so far discovered through GWAS collectively explain only a small fraction of the known heritability for any of the diseases [2–5].
Given what is known about the biology of these diseases and traits, it is suspected that important variants with moderate to large effect sizes remain to be identified; this is commonly referred to as the “missing heritability” [2, 3, 6, 7]. The explanations for missing heritability include the postulation that it could lie in regulatory rare variants, functional variants, structural variants, gene-by-gene or gene-by-environment interactions [8–11]. It has also been suggested that multiple small-effect variants, which are individually undetectable with the statistical power of GWAS, additively contribute to the missing heritability [12–14]. Another explanation is that the current estimates of total heritability may have been significantly inflated by the effects of epistasis [15]. The search for missing heritability has witnessed application of various approaches including pathway-based analysis of common, less frequent, and rare variants [16–18]; analysis of correlated traits using summary statistics from GWAS [19]; and analytical procedures that accommodate mixture of effects on the traits [20]. Because some of the existing effective pharmacotherapeutic agents for blood pressure control act by targeting specific biological pathways and these pathways are less represented in the GWAS-identified variants [1, 21–24], analytical approaches that focus on known biological pathways rather than on the entire genome could lead to discovery of some of the variants linked to “missing heritability” in association studies.
Consequently, the main objective of the present study was to perform pathway-based association analysis to identify blood pressure phenotypes–associated functional variants in the aldosterone-regulated sodium reabsorption pathway using whole exome sequence data provided for Genetic Analysis Workshop 19 (GAW19). The aldosterone pathway was chosen because it is one of the known target biological pathways for pharmacological control of hypertension. We hypothesize that functional genetic variant in the pathway influences susceptibility to blood pressure elevation.
Methods
Analyses were based on the unrelated data set of human whole exome sequence data plus the simulated and real phenotypes data as provided for GAW19 and described by Almasy et al [25].
Study subjects and phenotypes
The study samples included 1943 adult Hispanic subjects, that is, 1021 type 2 diabetes cases and 922 controls from the San Antonio Family Heart Study, San Antonio Family Diabetes/Gallbladder Study, Veterans Administration Genetic Epidemiology Study, and the Investigation of Nephropathy and Diabetes Study family component (HA) [26–29]; and the Starr County, Texas (HS) [30, 31] studies. Available study variables included sex, age, diastolic blood pressure (DBP), systolic blood pressure (SBP), and use of antihypertensive medication. Of the 1943 subjects, only 1850 had complete data on study variables.
We analyzed both the simulated blood pressure phenotypes in the “SIMPHEN.1” data set and the real blood pressure phenotypes in the “T2D-GENES_P1_Hispanic_phenotypes” data set. Outcome variables included in the analysis are DBP, SBP, pulse pressure (PP) (defined as PP = SBP − DBP), mean arterial pressure (MAP) (defined as MAP = DBP+[PP/3]), and hypertension (defined as blood pressure ≥140/90 mm Hg or use of antihypertensive medication). Sex, age, and age-squared were treated as covariates in the analysis.
Genotype data
Whole exome sequence data were provided on 11 odd-numbered autosomes. The genotypes used in the present analysis were based on NALTT (number of nonreference alleles for each individual thresholded) as provided in the variant call format (VCF) files. We used the software BCFtools (http://samtools.github.io/bcftools/bcftools.html) to extract data on biallelic (single nucleotide and deletion/insertion) variants and then recoded the genotypes from 0/1/2 to ACGT using the information on both the reference and alternate alleles for each variant. The quality control (QC) of the genotype data was carried out using the software PLINK [32]. Of the 1,765,688 total available variants, 1,711,766 were biallelic. We excluded 136,233 variants with missing genotypes greater than 10 % and 1,529,240 variants with minor allele frequency of less than 1 %. Rare variants were excluded because the focus of the analysis was on common and less-frequent variants and also because the sample size was too small for single-variant analysis of rare variants. Another 1238 variants that failed Hardy-Weinberg equilibrium test at p <0.001 were excluded. One sample with missing genotypes of greater than 10 % was excluded. The final quality-controlled genotype data set was made up of 1942 samples and 45,055 biallelic variants. Principal component analysis was performed using all 45,055 variants and the first of 10 components was extracted and included in association analysis to control for population stratification. Only the 1850 subjects with complete data on blood pressure phenotypes were included in association analysis.
Aldosterone-regulated sodium reabsorption pathway genes
The aldosterone-regulated sodium reabsorption pathway was defined using KEGG PATHWAY Database (http://www.genome.jp/kegg/pathway.html). The pathway comprises of 39 genes located across 14 autosomes and the X chromosome. Twenty-two genes were on the 11 odd-numbered autosomes available for the present analysis (Table 1). Annotation of variants was done using the SeattleSeq Annotation (http://snp.gs.washington.edu/SeattleSeqAnnotation138/index.jsp). Of the 45,055 biallelic variants that passed QC, a total of 127 were in the aldosterone-regulated sodium reabsorption pathway. With the exception of the SFN gene, each of the 22 genes on the odd-numbered autosomes had at least 1 variant available for analysis.
Association analysis
Using the simulated and real phenotypes, we fitted additive linear (for DBP, SBP, PP, MAP) and logistic (for hypertension status) regression models for each outcome variable with the variant as explanatory variable coded as dosage of the minor allele. Sex, age, age-squared, and first principal component were included as covariates. The software PLINK [32] was used for the association analysis by implementing the set-based tests. All the 127 variants in the pathway were considered as a set. The test involved iterative steps that included: (a) for each variant, we determined which other variants were in linkage disequilibrium above a certain threshold R 2 and eliminated other variants with values above the threshold; (b) performed single-variant association analysis and selected up to N variants with p values below P, starting with the most significant one; (c) from the subset of variants, we calculated set-statistic as the mean of the single-variant statistics; (d) permuted the data set 5000 times and repeated steps (b) and (c) for each permuted data set; (e) calculated empirical p value as the number of times the permuted set–statistic exceeded the original data set–statistic. Software default values of 0.5, 0.05, and 10 were used for the parameters R 2, P, and N, respectively. For each outcome variable, haplotype and epistasis association analyses were also done. The epistasis association involved all pairwise combinations of the 127 variants and their interaction. We also performed Bonferroni correction for multiple testing using number of testing as equal to the number of genes in the pathway. This was based on the assumption of nonindependence of variants in the pathway genes.
Results
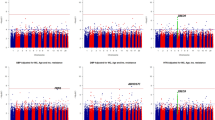
Figure 1 displays the distributions of the single-variant association analysis for both real and simulated phenotypes. Table 2 shows the topmost pathway-based association signals. After Bonferroni correction for multiple testing, associations of PRKCA gene with DBP, SBP, and MAP remained significant in both real and simulated data. None of the empirical p values reached significant level. Figure 2 displays the distributions of the haplotype associations. The haplotype signals are similar to those of the single variant analysis in Fig. 1. Table 3 shows the results of the 2-locus epistasis analysis. The most significant interactions for real phenotypes were those between different genes, for example, INS vs. PIK3R2 for DBP, whereas for simulated phenotypes there were significant within-genes interactions such as in INSR gene for SBP, MAP, and hypertension (Table 3).

Distributions of single single-nucleotide polymorphism association signals for real phenotypes (top) and simulated phenotypes (bottom)

Distributions of haplotype association signals for real phenotypes (top) and simulated phenotypes (bottom)
Discussion
In this study, we explored pathway-based analytical approach for the discovery of functional variants influencing blood phenotypes as additional method that could lead to identification of additional variants for complex human conditions. We focussed on a known biological pathway rather than pathways constructed from none proven biological systems. Our hypothesis was that because existing effective pharmacotherapeutic agents for blood pressure control act by targeting specific biological pathways, appropriate analytical methods that focus on such pathways could lead to identification of additional variants linked to complex human conditions than currently discovered by GWAS and candidate gene approaches. Results from this analysis indicate that, indeed, the use of known biological pathways for genetic association analysis can be a useful approach in the presence of true association since it takes advantage of the nonindependence of variants across pathway genes for setting threshold for statistical significance. We do note that because our analysis included genes from only the odd-numbered autosomes provided for GAW19, these results and their interpretations cannot be taken as fully representative of the aldosterone pathway. We are of the opinion that pathway-based analysis of variants from all genes in the pathway with those from the regulatory regions would lead to identification of important associations that can be interpreted with less limitation than in the present study. The use of known biological pathways in this study represents useful extension of genetic association analysis for complex human diseases.
Conclusions
The findings from this study show that pathway-based analytical approaches can be useful in identification of important disease-associated variants that are otherwise undetectable by GWAS because of the assumption of nonindependence of variants within and across pathway genes which leads to reduced penalty of multiple testing and thus less stringent statistical significance threshold.
References
A Catalog of Published Genome-Wide Association Studies. http://www.genome.gov/gwastudies
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747–53.
Maher B. Personal genomes: the case of the missing heritability. Nature. 2008;456(7218):18–21.
Hemminki K, Li X, Sundquist K, Sundquist J. Familial risks for common diseases: etiologic clues and guidance to gene identification. Mutat Res. 2008;658(3):247–58.
Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–78.
Hemminki K, Forsti A, Bermejo JL. The “common disease-common variant” hypothesis and familial risks. PLoS One. 2008;3(6):e2504.
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare variants create synthetic genome-wide associations. PLoS Biol. 2010;8(1):e1000294.
Eichler EE, Flint J, Gibson G, Kong A, Leal SM, Moore JH, Nadeau JH. Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet. 2010;11(6):446–50.
Visscher PM, Yang J, Goddard ME. A commentary on “common SNPs explain a large proportion of the heritability for human height” by Yang et al. Twin Res Hum Genet 2010. 2010;13(6):517–24.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42(7):565–9.
Pritchard JK. Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet. 2001;69(1):124–37.
Goldstein DB. Common genetic variation and human traits. N Engl J Med. 2009;360(17):1696–8.
Rockman MV. The QTN program and the alleles that matter for evolution: all that’s gold does not glitter. Evolution. 2012;66(1):1–17.
Visscher PM, Brown MA, McCarthy MI, Yang J. Five years of GWAS discovery. Am J Hum Genet. 2012;90(1):7–24.
Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109(4):1193–8.
Alsulami H, Liu X, Beyene J. Pathway-based analysis of rare and common variants to test for association with blood pressure. BMC Proc. 2014;8 Suppl 1:S101.
Edwards JS, Atlas SR, Wilson SM, Cooper CF, Luo L, Stidley CA. Integrated statistical and pathway approach to next-generation sequencing analysis: a family-based study of hypertension. BMC Proc. 2014;8 Suppl 1:S104.
Reder NP, Tayo BO, Salako B, Ogunniyi A, Adeyemo A, Rotimi C, Cooper RS. Adrenergic alpha-1 pathway is associated with hypertension among Nigerians in a pathway-focused analysis. PLoS One. 2012;7(5):e37145.
Zhu X, Feng T, Tayo BO, Liang J, Young JH, Franceschini N, Smith JA, Yanek LR, Sun YV, Edwards TL, et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am J Hum Genet. 2015;96(1):21–36.
Clarke GM, Rivas MA, Morris AP. A flexible approach for the analysis of rare variants allowing for a mixture of effects on binary or quantitative traits. PLoS Genet. 2013;9(8):e1003694.
Fox ER, Young JH, Li Y, Dreisbach AW, Keating BJ, Musani SK, Liu K, Morrison AC, Ganesh S, Kutlar A, et al. Association of genetic variation with systolic and diastolic blood pressure among African Americans: the Candidate Gene Association Resource study. Hum Mol Genet. 2011;20(11):2273–84.
Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, Smith AV, Tobin MD, Verwoert GC, Hwang SJ, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–9.
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–87.
Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41(6):666–76.
Blangero J, Teslovich TM, Sim X, Almeida MA, Jun G, Dyer TD, Johnson M, Peralta JM, Manning A, Wood AR, Fuchsberger C, Kent Jr JW, et al. Omics-squared: Human genomic, transcriptomic and phenotypic data for Genetic Analysis Workshop 19. BMC Proc. 2015;9 Suppl 8:S2.
Mitchell BD, Kammerer CM, Blangero J, Mahaney MC, Rainwater DL, Dyke B, Hixson JE, Henkel RD, Sharp RM, Comuzzie AG, et al. Genetic and environmental contributions to cardiovascular risk factors in Mexican Americans. The San Antonio Family Heart Study. Circulation. 1996;94(9):2159–70.
Hunt KJ, Lehman DM, Arya R, Fowler S, Leach RJ, Göring HH, Almasy L, Blangero J, Dyer TD, Duggirala R, et al. Genome-wide linkage analyses of type 2 diabetes in Mexican Americans: the San Antonio Family Diabetes/Gallbladder Study. Diabetes. 2005;54(9):2655–62.
Coletta DK, Schneider J, Hu SL, Dyer TD, Puppala S, Farook VS, Arya R, Lehman DM, Blangero J, DeFronzo RA, et al. Genome-wide linkage scan for genes influencing plasma triglyceride levels in the Veterans Administration Genetic Epidemiology Study. Diabetes. 2009;58(1):279–84.
Knowler WC, Coresh J, Elston RC, Freedman BI, Iyengar SK, Kimmel PL, Olson JM, Plaetke R, Sedor JR, Seldin MF. The Family Investigation of Nephropathy and Diabetes (FIND): design and methods. J Diabetes Complications. 2005;19(1):1–9.
Hanis CL, Ferrell RE, Barton SA, Aguilar L, Garza-Ibarra A, Tulloch BR, Garcia CA, Schull WJ. Diabetes among Mexican Americans in Starr County, Texas. Am J Epidemiol. 1983;118(5):659–72.
Below JE, Gamazon ER, Morrison JV, Konkashbaev A, Pluzhnikov A, McKeigue PM, Parra EJ, Elbein SC, Hallman DM, Nicolae DL, et al. Genome-wide association and meta-analysis in populations from Starr County, Texas, and Mexico City identify type 2 diabetes susceptibility loci and enrichment for expression quantitative trait loci in top signals. Diabetologia. 2011;54(8):2047–55.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Acknowledgements
This analysis was supported in part by grant HL53353 from the National Heart Lung and Blood Institute.
Declarations
This article has been published as part of BMC Proceedings Volume 10 Supplement 7, 2016: Genetic Analysis Workshop 19: Sequence, Blood Pressure and Expression Data. Summary articles. The full contents of the supplement are available online at http://bmcproc.biomedcentral.com/articles/supplements/volume-10-supplement-7. Publication of the proceedings of Genetic Analysis Workshop 19 was supported by National Institutes of Health grant R01 GM031575.
Authors’ contributions
BOT conceived of the study, performed all statistical analyses and wrote the manuscript. LT and RSC participated in the design and preparation of manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Tayo, B.O., Tong, L. & Cooper, R.S. Association of polymorphisms in the aldosterone-regulated sodium reabsorption pathway with blood pressure among Hispanics. BMC Proc 10 (Suppl 7), 15 (2016). https://doi.org/10.1186/s12919-016-0054-5
Published:
DOI: https://doi.org/10.1186/s12919-016-0054-5