
Abstract
Background
Meta-analysis has been widely used in genetic association studies to increase sample size and to improve power, both in the context of single-variant analysis, as well as for gene-based tests. Meta-analysis approaches for haplotype analysis have not been extensively developed and used, and have not been compared with other ways of jointly analysing multiple genetic variants.
Methods
We propose a novel meta-analysis approach for a gene-based haplotype association test, and compare it with an existing meta-analysis approach of the sequence kernel association test (SKAT), using the unrelated samples and family samples of the Genetic Analysis Workshop 19 data sets. We performed association tests with diastolic blood pressure and restricted our analyses to all variants in exonic regions on all odd chromosomes.
Results
Meta-analysis of haplotype results and SKAT identified different genes. The most significantly associated gene identified by SKAT was the ALCAM gene on chromosome 3 with a p value of 7.0 × 10− 5. Two of the most associated genes identified by the haplotype method were FPGT (p = 6.7 × 10− 8) on chromosome 1 and SPARC (p = 3.3 × 10− 7) on chromosome 5. Both genes were previously implicated in blood pressure regulation and hypertension.
Conclusion
We compared two meta-analysis approaches to jointly analyze multiple variants: SKAT and haplotype tests. The difference in observed results may be because the haplotype method considered all observed haplotypes, whereas SKAT weighted variants inversely to their minor allele frequency, masking the effects of common variants. The two approaches identified different top genes, and appear to be complementary.
Similar content being viewed by others


Background
In recent years, genome-wide association studies (GWAS) have unearthed a large number of single-nucleotide variants (SNVs) associated with many diseases [1]. Most associated variants have been common, with a minor allele frequency (MAF) of greater than 5 % and have been identified using a 1-SNV-at-a-time approach, where association with each SNV is evaluated separately, ignoring linkage disequilibrium (LD) between SNVs. A large number of associated variants were discovered only after combining GWAS results from multiple cohorts using meta-analysis approaches. To better focus on rare variants, methods to jointly analyse variants have been developed with meta-analysis extensions to combine results from multiple cohorts [2, 3]. These methods include burden tests, where association between a trait and the number of rare alleles a person carries is evaluated, and the sequence kernel association test (SKAT), which aggregates the evidence for association over multiple SNVs allowing for different direction of effects [4]. All of these approaches ignore the possibility of haplotype effects, where multiple alleles inherited together influence the trait. Even though single cohort approaches for haplotype analysis have been developed [5, 6], meta-analysis of haplotype results remains challenging because of the possibility of cohort-specific haplotype structure.
In this paper, we compare two multi-SNV analysis approaches applied to the Genetic Analysis Workshop 19 (GAW19) data, using both family and unrelated data. We use meta-analysis to combine the results from SKAT and haplotype analysis, using a novel approach for meta-analysis of haplotype results developed by our group. We apply these approaches to subsets of SNVs defined by gene location.
Methods
For all analyses considered, SNVs are grouped by gene location, and variants within each gene are tested for association with the phenotype of interest. Gene-based groupings were identified from the hg19 reference genome with the ANNOVAR (Annotate Variation) software [7]. From the family data set, we analyzed 464 individuals with sequence data available and 407 individuals from the unrelated data set.
We performed joint analysis of rare variants to assess association with diastolic blood pressure (DBP) adjusted for baseline age and sex. We adjusted DBP values for the use of blood pressure–lowering medication by adding ten to the observed DBP for all subjects reported to be on medication [8].
SKAT analyses were conducted with the RAREMETAL software [3]. Genome-wide single-variant association tests are calculated for family and unrelated samples separately with the RAREMETALWORKER software. The results were combined to calculate fixed-effect meta-analytic tests of association between the phenotype and groups of variants. The haplotype association test was implemented in R. Rare haplotypes (<0.5 %) were collapsed to ensure computational stability. To evaluate type 1 error rate of the haplotype analysis method, we analyzed genes on chromosome 17 from all 200 simulation replicates. We excluded genes located within 1 Mb of SNVs simulated to have an effect on any of the blood pressure traits.
Sequence kernel association test
The SKAT method [4] is based on a regression model of phenotype as a function of covariates and genotypes at all loci within a region. Familial relatedness was incorporated at the cohort level by means of the expected kinship matrix calculated from the pedigree structure. For a gene-based group containing p variants, with effect parameters β 1, ⋯, β p , the genotype-phenotype association is evaluated with the null hypothesis H0: β 1 = β 2 = ⋯ = β p = 0. Specifically, under the assumption that each β j is distributed with mean zero and variance w j τ where w j is a specified weight for SNV j, this is equivalent to the null hypothesis τ = 0, which is assessed by means of a variance components score test. In accordance with Wu et al. [4], weights are of the form w j = Beta(MAF j ; 1, 25)2 where the Beta distribution is evaluated at the MAF of that variant. This weights the rarest variants most heavily and smoothly reduces the weights of common variants, reflecting the assumption that natural selection against strong causal SNVs will result in lower frequency in the population. Several authors have developed approaches for meta-analysis of SKAT results, enabling the combination of GAW19 related and unrelated samples [2, 3].
Haplotype association test
We developed a novel approach to test the association between haplotype structure and phenotype so as to better understand the genetic architecture of each region and its influence on DBP. We incorporated family structure into the model proposed by Zaykin et al. [6] so that our approach is applicable to both unrelated and related samples. For K observed haplotypes, we model the phenotype at the cohort level as
where Y is the trait (DBP at baseline), X are covariates such as age and sex with no intercept, h k is the dosage of the k th haplotype out of K observed haplotypes, b ~ N(0, 2σ 2Σ kin ) is a random effect vector that accounts for the familial correlation, Σ kin is the expected kinship matrix derived from the pedigree and ε is the residual error. In our implementation, haplotype dosages are estimated from genotypes using an expectation–maximization algorithm (R Package haplo.stats [5]) ignoring familial information but exploiting the LD among genetic variants. We use a weighted least-squares method [9] to meta-analyze the beta coefficients for the K′ haplotypes observed in one or more cohorts. After meta-analysis, we test the global null hypothesis that all haplotype effects are equal (ie, H0: β 1 = ⋯ = β K '). A Wald test with df = K′ − 1 is implemented to test the reparameterized null hypothesis H0: γ 2 = ⋯ = γ K ' = 0 where γ i = β i − β c and the subscript “C” refers to one of the haplotypes common to all studies selected at random.
Results
Both methods require specification of SNV sets for combined analysis. We restrict our attention to variants in exonic regions, and group SNVs by genes. We considered 8806 genes across the odd-numbered chromosomes. Of these, 135 contained only one SNV, while the obscurin protein-coding (OBSCN) gene on chromosome 1 had the maximum of 1054 SNVs in its exonic regions. Figure 1 shows the distribution of SNVs per gene.

Number of SNVs with a MAF <5 % for each gene grouping

Association results for all odd chromosomes: the upper half represents SKAT -log10(p value); the lower half shows log10(p value) for the haplotype method
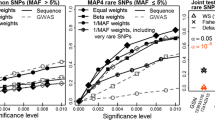
Type I error evaluation of the novel haplotype association method revealed an inflated type I error rate for genes with more than 14 haplotypes, but not for genes with fewer haplotypes (type I error rate = 0.053 at α = 0.05, 95 % confidence interval from 0.0495 to 0.0567). Hence, in Fig. 2 we report only results from genes with fewer than 14 haplotypes, after collapsing rare haplotypes together.
The most significant gene-based SKAT result was found in the activated leukocyte cell adhesion molecule (ALCAM) gene on chromosome 3; Table 1 lists the top ten SKAT association results. This gene codes for an immunoglobulin protein that is expressed in neural and epithelial cells [10]. The association p value with the set of 28 ALCAM SNVs was 7.0 × 10−5. However, none of the SKAT gene-based tests reached the genome-wide significance threshold, with Bonferroni correction for the 8806 genes tested (5.7 × 10−6).
Two of the most significant genes found to be associated with DBP by the haplotype method were fucose-1-phosphate guanylyltransferase (FPGT) on chromosome 1 and secreted protein, acidic, cysteine-rich (SPARC) on chromosome 5. Both genes (p FPGT = 6.7 × 10− 8; p SPARC = 3.3 × 10− 7) reached the genome-wide significance using Bonferoni correction. Table 2 lists the top ten haplotype association results.
Discussion
The top gene association from the SKAT analysis, ALCAM, has been identified in a quantitative trait locus for systolic blood pressure in previous literature [11] and has shown differential gene expression in rats with hypertension [12].
The meta-analysis of haplotype results identified as its second strongest signal the gene FPGT, a gene in LD with a single nucleotide polymorphism (SNP) previously reported to be associated (p = 7.2 × 10− 5) with DBP in the lymphoblastoid cell line [13]. Our haplotype methods detected a much stronger association between FPGT and DBP (p = 6.7 × 10− 8). This stronger signal might be the result of the joint impact of all the five SNVs located in this gene. The sixth most significant gene identified by the haplotype approach, SPARC, contains three variants and was previously shown to be associated with cardiac dysfunction [14].
Conclusions
We performed gene-based multi-SNV analyses to identify regions of the genome associated with DBP. While we observed some consistencies between the SKAT and haplotype analyses, the haplotype analysis revealed multiple genome-wide significant results, some in genes that have been previously implicated in blood pressure regulation. Further investigation in a larger number of participants is needed to confirm the novel associations identified in this report.
References
Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42(Database issue):D1001–6.
Lee S, Teslovich TM, Boehnke M, Lin X. General framework for meta-analysis of rare variants in sequencing association studies. Am J Hum Genet. 2013;93(1):42–53.
Feng S, Liu D, Zhan X, Wing MK, Abecasis GR. RAREMETAL: fast and powerful meta-analysis for rare variants. Bioinformatics. 2014;30(19):2828–9.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89(1):82–93.
Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70(2):425–34.
Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG. Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered. 2002;53(2):79–91.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Tobin MD, Sheehan NA, Scurrah KJ, Burton PR. Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med. 2005;24(19):2911–35.
Becker BJ, Wu M. The synthesis of regression slopes in meta-analysis. Stat Sci. 2007;22(3):414–29.
Bowen MA, Patel DD, Li X, Modrell B, Malacko AR, Wang WC, Marquardt H, Neubauer M, Pesando JM, Francke U, et al. Cloning, mapping, and characterization of activated leukocyte-cell adhesion molecule (ALCAM), a CD6 ligand. J Exp Med. 1995;181(6):2213–20.
Rice T, Rankinen T, Province MA, Chagnon YC, Perusse L, Borecki IB, Bouchard C, Rao DC. Genome-wide linkage analysis of systolic and diastolic blood pressure: the Quebec Family Study. Circulation. 2000;102(16):1956–63.
Marques FZ, Campain AE, Yang YH, Morris BJ. Meta-analysis of genome-wide gene expression differences in onset and maintenance phases of genetic hypertension. Hypertension. 2010;56(2):319–24.
Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, Glazer NL, Morrison AC, Johnson AD, Aspelund T, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–87.
Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, d'Hooge J, Van de Werf F, Carmeliet P, Pinto YM, et al. Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med. 2009;206(1):113–23.
Acknowledgements
We want to acknowledge Achilleas Pitsillides for his computing assistance, and Honghuang Lin and Jaeyoung Hong for help with the annotation.
Declarations
This article has been published as part of BMC Proceedings Volume 10 Supplement 7, 2016: Genetic Analysis Workshop 19: Sequence, Blood Pressure and Expression Data. Summary articles. The full contents of the supplement are available online at http://bmcproc.biomedcentral.com/articles/supplements/volume-10-supplement-7. Publication of the proceedings of Genetic Analysis Workshop 19 was supported by National Institutes of Health grant R01 GM031575.
Authors’ contributions
JD conceived of the project and provided critical revision to the manuscript. SW and VAF drafted the manuscript and performed statistical analyses. YC performed statistical analyses and provided critical revision to the manuscript. SW and VF contributed equally to this work. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, S., Fisher, V.A., Chen, Y. et al. Comparison of multiple single-nucleotide variant association tests in a meta-analysis of Genetic Analysis Workshop 19 family and unrelated data. BMC Proc 10 (Suppl 7), 51 (2016). https://doi.org/10.1186/s12919-016-0028-7
Published:
DOI: https://doi.org/10.1186/s12919-016-0028-7