
Abstract
Background
Building the evolutionary trees for massive unaligned DNA sequences is challenging and crucial. However, reconstructing evolutionary tree for ultra-large sequences is hard. Massive multiple sequence alignment is also challenging and time/space consuming. Hadoop and Spark are developed recently, which bring spring light for the classical computational biology problems. In this paper, we tried to solve the multiple sequence alignment and evolutionary reconstruction in parallel.
Results
HPTree, which is developed in this paper, can deal with big DNA sequence files quickly. It works well on the >1GB files, and gets better performance than other evolutionary reconstruction tools. Users could use HPTree for reonstructing evolutioanry trees on the computer clusters or cloud platform (eg. Amazon Cloud). HPTree could help on population evolution research and metagenomics analysis.
Conclusions
In this paper, we employ the Hadoop and Spark platform and design an evolutionary tree reconstruction software tool for unaligned massive DNA sequences. Clustering and multiple sequence alignment are done in parallel. Neighbour-joining model was employed for the evolutionary tree building. We opened our software together with source codes via http://lab.malab.cn/soft/HPtree/.
Similar content being viewed by others


Background
The reconstruction of evolution trees and alignments for large data are still open challenges for bioinformatics researchers [1, 2]. The third generation sequencing techniques promoted massive metagenome sequences, which call for OTU clustering and taxonomic labelling [3]. Besides deep understanding on the genes, population, species evolutionary relationships, evolutionary tree reconstruction also benefits for the metagenome and microbial genomics research.
Evolutionary tree reconstruction could be divided into three different situations. The first one is different species genomes evolutionary relationship reconstruction, which considers the influence from horizontal gene transfer [4, 5], incomplete lineage sorting [6, 7], gene orders with insertions and deletions [8], and rearrangement [9]. The second situation considers different homologous genes [10], where the maximum likelihood method is usually chosen for their perfect mathematical explanation [11]. Some researchers considered that networks could represent the evolutionary process better than trees [12]. The third one is to analyze the evolutionary relationships among the individuals in a population. In this case, massive similar sequences should be handled, and computer memory limitation often becomes the bottleneck.
Multiple sequence alignment is necessary for evolutionary tree software tools, including MEGA [13], MAFFT [14], SATe-II [15], IQ-TREE [16], iGTP [17], FastTree [18], and phangorn [19]. Most multiple sequence alignment tools cannot deal with massive sequences (eg. >10,000 sequences). Therefore, evolutionary tree reconstruction independent of multiple sequence alignment was developed [20], which is called the next-generation phylogenomics [21]. The divide-and-conquer algorithm [22] and distance model [23] have been employed, and the sequence distance was be computed according to different types of kmers [24, 25], word frequencies [26] or average common substrings [27]. They can avoid the time cost from pairwise sequence alignment. However, the performance would be decreased [28]. Due to the lack of evolutionary reconstruction tools together with multiple sequence alignment for massive unaligned sequences, it is essential and necessary to solve this problem with latest parallel computation techniques.
Some parallel techniques were tested for evolutionary tree, including multi-cores [29, 30], MPI [31,32,33], grid computing [34], GPU [35, 36], etc. However, there are no related references on the Hadoop and Spark platform. Hadoop has been utilized in multiple sequence alignment for handling large scale data in our previous work [37]. Here we build the evolutionary tree for massive unaligned DNA sequences with Hadoop and Spark framework.
Results
Data
TreeBase [38] was selected as the golden benchmark in most of the current evolutioanry reconstruction software tools. But the data from TreeBase are rather small. We try to solve the massive sequences problem, so TreeBase is not suitable in this work. Since there is no large scale benchmark datasets, we only selected running time as the performance measurement.
Human mitochondrial genomes [39] and 16S rRNAs [40] were employed for testing in our work. There are 672 human mitochondrial genomes in the human mitochondrial genomes dataset. In order to test the “big data” performance, the data were duplicated 20, 50, and 100 times separately. In these datasets, sequences were similar. We also tested the performance in the 16S rRNAs datasets, in which sequences have low similarity. There are two differnt files. The first file is 156 MB, while the second is 1.4 GB. All the datasets seemed more bigger than TreeBase.
Comparison with the state-of-the-art software tools
We have tried our datasets with MEGA [13], MAFFT [14], SATe-II [15], RAxML [33], STELLS [41], MrBayes [30], Beagle [42], Beast [43], and PLL [31]. However, most of them cannot even handle the smallest dataset. So we only compare the performance with RAxML, Phangorn, STELLS and IQ-Tree [16]. All these three software tools need the aligned files as the input. So we firstly emloyed HAlign [44] for multiple sequence alignment before the evolutionary trees reconstruction.
The running time was compared and showed in Fig. 1. The initial human mitochondrial genomes dataset is about 10 MB. After the duplicating, the 100× file is more than 1 GB. Since other tools can only work on single node, all the software tools were testing in one computer instead of the cluster. In Fig. 1, it seemed that HPTree outperform other tools even on a single node. Phangorn, RAxML and STELLS could handle the 1× file but cannot deal with the larger files.

The running time on mt genome datasets with different numbers of Hadoop nodes. Running time of different software tools on mtDNA datasets
For the massive high-similarity DNA sequences, HPTree can build the evolutionary tree for over 1 GB file in several minutes. Then we tested the performance with low-similarity sequences. In our testing experiments, only HPTree could handle the two 16S rRNA datasets. The consuming time was shown in Fig. 2. For the low-similarity datasets, HPTree still works for the more than 1GB files.

The running time on mt genome datasets with different numbers of Spark nodes. Running time with HPTree on 16S rRNA datasets
Speed up of the parallel mechanism
Single node performance of HPTree was shown in Figs. 1 and 2. Here we employed Hadoop on 2–4 nodes, and showed the performance of HPTree in clusters. Table 1 showed the running time of different nodes comparison. The parallel efficiency and the speed-up ratio of HPTree with Hadoop and Spark can be viewed from Table 1. The multi-codes cluster would accelerate and benefit for the massive big files. Besides, we can see that Spark platform has a better average performance than native Hadoop, which owes to the memory computing technology in Spark. For Hadoop MapReduce operator, all intermediate data will be saved in hard disk for disaster recovery, which is suitable for massive data processing but reduces the efficiency of programs. For Spark platform, intermediate data will be saved in memory as much as possible for reiterative computing, and the rest of intermediate data will be saved in hard disk [45]. Hence, our experiment result shows that Spark accelerate HPTree more remarkably than Hadoop.
Performance on the unaligned sequences
We have employed Halign for multiple sequence alignment as preprocess in the above testing. The most important point of HPTree is the ability of handling unaligned sequences, which is the key advantage beyond other evolutionary reconstruction tools. Multiple sequence alignment for massive sequences is also challenging and time/space consuming. HPTree also deals with this problem in parallel.
Tables 2 and 3 shows the running time of aligned and unaligned data on Hadoop and Spark platform, respectively. Our bigdata sets, including human mitochondrial genomes and 16S rRNAs, were both tested. Because no other software tool could deal with the unaligned sequences, we just show the expresiment results of HPTree. Tables 2 and 3 showed several interesting results. We conclude that HPTree runs observably faster on Spark platform than on Hadoop platform. As the sequence number grows larger, the multiple sequence alignment would occupy littler in the total running time. In the 1× and 20× datasets, running time increased sharply for the unaligned sequences. But in the 50×, 100× and 16 s rRNA datasets, which contain more than 10, 000 sequences, multiple sequence alignment would not influence the time performance sharply. Neighbour joining occupied most of the running time. It suggests that multiple sequence alignment is not the only problem for massive sequences, but the tree topology and branch distance computation is also the key challenge.
Comparison with HPTree on Hadoop and spark
Hadoop and Spark are popular distributed computing frameworks. Fault-tolerant for the former relied on HDFS system based on backups on hard disk. Such a design is suitable for ultra-large dataset (larger than TB class) because memory is not able to load such ultra-large dataset. Facts proved that Hadoop framework has achieved great success on the distributed computing field based on MapReduce programming model. Fault-tolerant for Spark relied on RDD data structure based on backups on memory and hard disk. From the above experiment result, efficient reiterative computing on HPTree for Spark runs faster than HPTree on Hadoop. However, Spark is closely associated with Hadoop, and HDFS system would not be replaced. Ultra-large dataset need to be persisted on Hadoop HDFS system based on hard disk and need to be efficient computed on Spark MapReduce framework. Our experiments show that Spark and HDFS system can cope with ultra-large multiple sequence alignment and evolutionary analysis issue.
Discussion
Evolutionary tree reconstruction for massive unaligned sequences is still an open challenge. In this paper, we employed the Hadoop and Spark platform for the massive DNA sequences evolutionary relationship analysis in parallel. In order to decrease the running time, the neighbour-joining model is chosen for the tree building. Different from the common neighbour-joining algorithm, sequence clustering is done first and multiple sequence alignment and sub-evolutionary tree construction are excuted in parallel on each node. The final tree is built combining the subtree results. HPTree could handle the unaligned massive sequences, while the state-of-arts tools cannot. In the more than 1 GB files experiments, HPTree works well on both high and low similarity sequences.
In this work, edit distance is chosed to measure the evolutionary distance because of simplicity and speed. Indeed, more complex evolutionary distance models should be considered, which is the future work. A smarter data structure on edit distance [46, 47] would facilitate the acceleration of parallel computing, which is also an opportunity for future work and improvement.
RNA is viewed similarly to DNA in this work. The RNA secondary structure information [48,49,50,51] is not considered in the alignment or involved in the evolutionary tree built by HPTree. RNA clustering or evolutionary analysis always requires secondary structure or base pair-matching information, such as microRNA family annotation in the miRBase [52] and RFam database [53, 54]. Therefore, a Hidden Markov Model is always employed in the alignment and distance computing process, but it is rather time consuming and unsuitable for ultra-large data.
Moreover, evolutionary networks are superior to trees for large-scale and complex evolutionary analysis. Our parallel strategy also suits network reconstruction. However, this approach is somewhat complicated and will be undertaken in the future.
Methods
Although MapReduce frame was employed in HPTree similar to multiple sequence alignment, the core techniques were totally different from HAlign. The main core problem is the subtree partition, which involved the load balancing in each node in the next step. Here we also employed MapReduce to clustering the massive sequences firstly. If some clusters are more bigger or smaller than others, we will split the big clusters and combine the small ones. Related techniques were introduced in our previous work [55, 56].
After clustering, we chose neighbour-joining (NJ) model for the subtree construction. Comparing with maximum parsimony and maximum likelihood models, neighbour-joining is fast and least time/space consuming. Maximum parsimony and maximum likelihood models are both complex and not suitable for the MapReduce frame.
The major advantage of HPTree is the ability for unaligned sequences. We employed Hadoop and Spark for the multiple sequence alignment in the preprocess. Then we introduced the detailed process respectively.
Multiple sequence alignment with Hadoop
We aimed at the ultra-large scale data. So in every step we selected the simplest model and method. Here center star multiple sequence alignment algorithm was chosen instead of tree based alignment algorithm. In center star algorithm, “centre sequence” is chosen as a standard one, and every other sequence would be aligned with the “centre sequence” pairwisely. After the pairwise alignments, all the spaces inserted to the “centre sequence” would be summed up, and the other sequences will be supplied the corresponding spaces. Finally, all the sequences will have the same length. This is the whole process of the center star multiple sequence alignment.
We employ Hadoop to accelerate this process in parallel. So all the sequences would be divided into several parts. In order to save time, we randomly selected a sequence as the “centre sequence”.
It is known that the entries in Map Reduce are recorded using the (key, value) format. We use key to denote the sequence name and value for the DNA sequence. Before the parallel computing, we pre-process the input sequences and delete the illegal characters and strange sequences. Then, all the input sequences are formatted as (key, value) pairs for Hadoop. As all the sequences are similar, the first sequence is selected as the centre sequence.
In the first stage of the Map function, the data file is divided automatically into several split files of size 64 MB or less. These split files are sent to different data nodes and aligned to the centre sequence in parallel. After the alignment, the centre sequence and the sequence in the split file are updated with inserted spaces. They are still recorded using the (key, value) format, where the key is the sequence name and the value is the updated two aligned sequences. Then, the output (key, value) pairs enter the Reduce stage.
In the first stage of the Reduce function, data are not processed and are output to the HDFS file system directly. The data are then collected from the HDFS file system to a local computer, and the aligned centre sequences are extracted and gathered. For the n aligned centre sequences, we count the maximum spaces between every two neighbouring characters. The maximum spaces are retained for the Final Centre Sequence.
The second Map-Reduce phase is similar to the first stage. All the aligned sequences in the first stage are aligned again to the Final Centre Sequence. As the Final Centre Sequence has the maximum spaces between all characters, there will be no spaces inserted into the Final Centre Sequence. Thus, all the other sequences will be aligned to the same length with the Final Centre Sequence, producing the final alignment result. Indeed, the original centre star method records the inserted space positions for the Final Centre Sequence instead of the second alignment. However, when handling massive data, the distributed storage of the records is a problem. As the DNA sequences are similar, the k-band alignment is linearly time consuming. Thus, the second Map-Reduce alignment is employed.
Multiple sequence alignment with spark
Hadoop mainly contains Hadoop Distributed File System (HDFS) for distributed storage and MapReduce programming model for big datasets. HDFS stores data on inexpensive machines, providing dependable fault-tolerant mechanism and high-aggregate bandwidth across clusters. Spark aims to blueprint a programming model that extends applications of MapReduce model and achieves high computational efficiency-based memory cache.
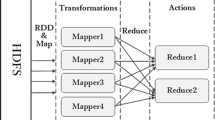
Spark designs an abstract data structure named resilient distributed datasets (RDDs) to support efficient computing and to ensure distribution of datasets on cluster machines. RDDs staying in memory cache will visibly reduce load time when requiring replication, especially in iterative operations. From Fig. 3, to further reduce time and cost, two types of operations in RDDs are designed: transforms and actions. Transforms only deliver computing graphs, which only describe how to compute and not how to carry out computing operations, such as map and filter operation. Actions carry out computing, such as reduce and collect operations, results of which are stored as new RDDs. Based on these operations, RDDs are efficiently executed in parallel. To ensure dependable fault tolerance, RDDs will be recomputed after data loss, for example, because of halting of individual machines. Based on RDDs, Spark can implement up to 100 times theoretical speed than Hadoop in real-world datasets.

MSA procedures based on Spark distributed framework
As mentioned before, based on parallel computing, we first cluster all MSA results into several clusters. Then, we calculate individual phylogenetic tree based on individual clusters. Last, all phylogenetic trees are merged on clusters into the final evolution tree. The approach comprises two key steps: initial clustering and MSA. MSA methods are determined by trie trees algorithm for similar nucleotide sequences. Then, we highlight the initial clustering procedure. Approximately 10% of all sequences are selected by random sampling for initial clustering. Then, functional distance of each pairwise sequence is calculated, clustered, and labeled until all sequences are identified. When few clusters whose number of elements is over 10%, then they are merged into other clusters; otherwise, they are divided into more balanced clusters until balanced construction. The entire procedure is designed for Spark parallel model.
Implementation
HPTree is licensed under the GPL license and is implemented using Java, which can work on multiple operation systems. Hadoop 2.0 and Spark 2.0 are required for the parallel tool. We have constructed the web site http://lab.malab.cn/soft/HPtree/ for sharing the data, codes and software tools. A friendly web server is also developed. Users with just internet browser could draw the evolutionary trees by uploading their zipped fasta files.
Conclusions
In this paper, we accelerate the evolutionary tree reconstruction with Hadoop and Spark. The ability of handling big data is also improved, especially unaligned sequences would be dealt with. Besides evolutionary analysis, the tree would also benefit for several other applications, such as DNA/protein sequence representation [57].
It can be anticipated that the proposed computational pipeline will have many potential applications. The multiple sequence alignment is one of the key techniques in biological sequence analysis. The proposed methods are able to efficiently reduce the computational cost, and therefore, they would be applied to protein, RNA, and DNA sequence analysis [58]. Recently, some algorithms have been proposed to extraction the evolutionary information from multiple sequence alignments, such as Pse-Analysis [59], and pseudo proteins [60]. Future studies will focus on extracting features from the evolutionary information.
References
Liu K, Raghavan S, Nelesen S, Linder CR, Warnow T. Rapid and accurate large-scale coestimation of sequence alignments and phylogenetic trees. Science. 2009;324(5934):1561–4.
Liu B, Zhang D, Xu R, Xu J, Wang X, Chen Q, Dong Q, Chou K-C. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics. 2014;30(4):472–9.
Segata N, Börnigen D, Morgan XC, Huttenhower C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat Commun. 2013;4(4):2304.
Lapierre P, Laseknesselquist E, Gogarten JP. The impact of HGT on phylogenomic reconstruction methods. Brief Bioinform. 2014;15(1):79–90.
Weyenberg G, Huggins PM, Schardl CL, Howe DK, Yoshida R. kdetrees: non-parametric estimation of phylogenetic tree distributions. Bioinformatics. 2014;30(16):2280–7.
Bayzid MS, Hunt T, Warnow T. Disk covering methods improve phylogenomic analyses. BMC Genomics. 2014;15(S6):S7.
Ané C. Detecting Phylogenetic breakpoints and discordance from genome-wide alignments for species tree reconstruction. Genome Biol Evol. 2011;3(3):246–58.
Hu F, Zhou J, Zhou L, Tang J. Probabilistic reconstruction of ancestral gene orders with insertions and deletions. IEEE/ACM Trans Comput Biol Bioinform. 2014;11(4):667–72.
Doyon JP, Ranwez V, Daubin V, Berry V. Models, algorithms and programs for phylogeny reconciliation. Brief Bioinform. 2011;12(5):392–400.
Chesters D, Zheng WM, Zhu CD. A DNA Barcoding system integrating multigene sequence data. Methods Ecol Evol. 2015;6(8):930–7.
Breinholt JW, Kawahara AY. Phylotranscriptomics: saturated third codon positions radically influence the estimation of trees based on next-gen data. Genome Biol Evol. 2013;5(11):2082–92.
Wang J, Guo M, Liu X, Liu Y, Wang C, Xing L, Che K. LNETWORK: an efficient and effective method for constructing phylogenetic networks. Bioinformatics. 2013;29(18):2269–76.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9.
Katoh K, Toh H. PartTree: an algorithm to build an approximate tree from a large number of unaligned sequences. Bioinformatics. 2007;23(3):372.
Liu K, Warnow TJ, Holder MT, Nelesen SM, Yu J, Stamatakis AP, Linder CR. SATe-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees. Syst Biol. 2012;61(1):90–106.
Nguyen LT, Schmidt HA, Haeseler AV, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32(1):268–74.
David FB, André W, Bansal MS, Ruchi C, Oliver E. iGTP: a software package for large-scale gene tree parsimony analysis. BMC Bioinformatics. 2010;11(1):574.
Price MN, Dehal PS, Arkin AP. FastTree 2 – approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(5):e9490.
Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. 2013;27(4):592–3.
Schwende I, Pham TD. Pattern recognition and probabilistic measures in alignment-free sequence analysis. Brief Bioinform. 2014;15(3):354.
Chan CX, Ragan MA. Next-generation phylogenomics. Biol Direct. 2013;8(1):3.
Nelesen S, Liu K, Wang LS, Linder CR, Warnow T. DACTAL: divide-and-conquer trees (almost) without alignments. Bioinformatics. 2012;28(12):274–82.
Chan CX, Bernard G, Poirion O, Hogan JM, Ragan MA. Inferring phylogenies of evolving sequences without multiple sequence alignment. Sci Rep. 2014;4(39):6504.
Tran NH, Chen X. Comparison of next-generation sequencing samples using compression-based distances and its application to phylogenetic reconstruction. BMC Res Notes. 2014;7(1):1–13.
Horwege S, Lindner S, Boden M, Hatje K, Kollmar M, Leimeister CA, Morgenstern B. Spaced words and kmacs: fast alignment-free sequence comparison based on inexact word matches. Nucleic Acids Res. 2014;42(Web Server issue):7–11.
Leimeister CA, Boden M, Horwege S, Lindner S, Morgenstern B. Fast alignment-free sequence comparison using spaced-word frequencies. Bioinformatics. 2014;30(14):1991–9.
Leimeister C-A, Morgenstern B. Kmacs: the k-mismatch average common substring approach to alignment-free sequence comparison. Bioinformatics. 2014;30(14):2000–8.
Tan G, Muffato M, Ledergerber C, Herrero J, Goldman N, Gil M, Dessimoz C. Current methods for automated filtering of multiple sequence alignments frequently worsen single-gene phylogenetic inference. Syst Biol. 2015;64(5):778–91.
Darriba D, Taboada GL, Doallo R, Posada D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics. 2011;27(8):1164–5.
Ronquist F, Teslenko M, PVD M, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539.
Flouri T, Izquierdo-Carrasco F, Darriba D, Aberer AJ, Nguyen LT, Minh BQ, Von HA, Stamatakis A. The phylogenetic likelihood library. Syst Biol. 2015;64(2):356–62.
Lartillot N, Rodrigue N, Stubbs D, Richer J. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst Biol. 2013;62(4):611.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312.
Bazinet AL, Zwickl DJ, Cummings MP. A gateway for phylogenetic analysis powered by grid computing featuring GARLI 2.0. Syst Biol. 2014;63(5):812–8.
Ayres DL, Darling A, Zwickl DJ, Beerli P, Holder MT, Lewis PO, Huelsenbeck JP, Ronquist F, Swofford DL, Cummings MP. BEAGLE: an application programming interface and high-performance computing library for statistical phylogenetics. Syst Biol. 2012;61(1):170–3.
Chen X, Wang C, Tang S, Yu C, Zou Q. CMSA: a heterogeneous CPU/GPU computing system for multiple similar RNA/DNA sequence alignment. BMC Bioinformatics. 2017;18:315.
Zou Q, Li XB, Jiang WR, Lin ZY, Li GL, Chen K. Survey of MapReduce frame operation in bioinformatics. Brief Bioinform. 2014;15(4):637.
Morell V. The roots of phylogeny. Science. 1996;273(5275):569.
Tanaka M, Cabrera VM, González AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, Hirose R, Fujita Y, Kurata M. Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res. 2004;14(10A):1832.
Jr DST, Hugenholtz P, Keller K, Brodie EL, Larsen N, Piceno YM, Phan R, Andersen GL. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res. 2006;34(2):394–9.
Wu Y. Coalescent-based species tree inference from gene tree topologies under incomplete lineage sorting by maximum likelihood. Evolution. 2012;66(3):763–75.
Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am J Hum Genet. 2007;81(5):1084–97.
Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–73.
Zou Q, Hu Q, Guo M, Wang G. HAlign: fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics. 2015;31(15):2475–81.
Shanahan JG, Dai L. Large Scale Distributed Data Science Using Apache Spark. In: ACM SIGKDD International Conference on Knowledge Discovery and Data Mining; 2015. p. 2323–4.
Wang J, Li G, Feng J. Extending string similarity join to tolerant fuzzy token matching 2014, 39(1):1-45.
Li G, Deng D, Feng J. A partition-based method for string similarity joins with edit-distance constraints. ACM Trans Database Syst. 2013;38(2):1–33.
Zou Q, Lin C, Liu XY, Han YP, Li WB, Guo MZ. Novel representation of RNA secondary structure used to improve prediction algorithms. Genet Mol Res. 2011;10(3):1986–98.
Zou Q, Zhao T, Liu Y, Guo M. Predicting RNA secondary structure based on the class information and Hopfield network. Comput Biol Med. 2009;39(3):206–14.
Liu B, Fang L, Liu F, Wang X, Chen J, Chou K-C. Identification of real microRNA precursors with a pseudo structure status composition approach. PLoS One. 2015;10(3):e0121501.
Liu B, Liu F, Fang L, Wang X, Chou K-C. repRNA: a web server for generating various feature vectors of RNA sequences. Mol Gen Genomics. 2016;291(1):473–81.
Kozomara A, Griffithsjones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42(1):68–73.
Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res. 2013;41(1):226–32.
Huang Y, Liu N, Wang JP, Wang YQ, Yu XL, Wang ZB, Cheng XC, Zou Q: Regulatory long non-coding RNA and its functions. J Physiol Biochem 2012, 68(4):611-618.
Zou Q, Wan S, Zeng X. HPTree: Reconstructing phylogenetic trees for ultra-large unaligned DNA sequences via NJ model and Hadoop. In: IEEE International Conference on Bioinformatics and Biomedicine; 2017. p. 53–8.
Zou Q. Multiple sequence alignment and reconstructing phylogenetic trees with Hadoop. In: IEEE International Conference on Bioinformatics and Biomedicine; 2017.
Wei L, Tang J, Zou Q. Local-DPP: an improved DNA-binding protein prediction method by exploring local evolutionary information. Inf Sci. 2017;384:135–44.
Liu B, Liu F, Wang X, Chen J, Fang L, Chou K-C. Pse-in-one: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015;43(W1):W65–71.
Liu B, Wu H, Wang X, Chou K-C. Pse-analysis a python package for DNA, RNA and protein peptide sequence analysis based on pseudo components and kernel methods. Oncotarget. 2017;8(8):13338–43.
Chen J, Long R, Wang X, Liu B, Chou K-C. dRHP-PseRA: detecting remote homology proteins using profile based pseudo protein sequence and rank aggregation. Sci Rep. 2016;6:32333.
Acknowledgements
Not Applicable.
Funding
Publication costs were funded by the Natural Science Foundation of China (No. 61771331).
Availability of data and materials
All data generated and analyzed during this study are included in this published article (mentioned in the section “Methods”) and the web sites.
About this supplement
This article has been published as part of BMC Systems Biology Volume 11 Supplement 6, 2017: Selected articles from the IEEE BIBM International Conference on Bioinformatics & Biomedicine (BIBM) 2016: systems biology. The full contents of the supplement are available online at https://bmcsystbiol.biomedcentral.com/articles/supplements/volume-11-supplement-6.
Author information
Authors and Affiliations
Contributions
QZ wrote the manuscript and coordinated the project. SW helped to do the experiments on Spark. XZ helped to revise the manuscript and do the experiments on Hadoop. ZM gave helpful suggestions and helped to revise the English. All of the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zou, Q., Wan, S., Zeng, X. et al. Reconstructing evolutionary trees in parallel for massive sequences. BMC Syst Biol 11 (Suppl 6), 100 (2017). https://doi.org/10.1186/s12918-017-0476-3
Published:
DOI: https://doi.org/10.1186/s12918-017-0476-3