
Abstract
Background
Aging is a complex process relating multi-scale omics data. Finding key age markers in normal tissues could help to provide reliable aging predictions in human. However, predicting age based on multi-omics data with both accuracy and informative biological function has not been performed systematically, thus relative cross-tissue analysis has not been investigated entirely, either.
Results
Here we have developed an improved prediction pipeline, the Integrating and Stepwise Age-Prediction (ISAP) method, to regress age and find key aging markers effectively. Furthermore, we have performed a serious of network analyses, such as the PPI network, cross-tissue networks and pathway interaction networks.
Conclusion
Our results find important coordinated aging patterns between different tissues. Both co-profiling and cross-pathway analyses identify more thorough functions of aging, and could help to find aging markers, pathways and relative aging disease researches.
Similar content being viewed by others

Background
Aging is a multi-faceted and progressive bio-process for many organisms [1]. The aging process is composed by a serious of complex dynamic molecular interactions [2], which indicate key physiological phenotypes of human health. Moreover, dysfunctions of aging process have been shown to be involved in many disorders such as Parkinson disease [3], Alzheimers’ disease [4], many kinds of cancers [5] and so on. Therefore finding important aging markers could provide opportunities to predict healthy factors and improve diagnostic results for both pre- and pro- gnosis [6].
It has been reported that profiling patterns of crucial DNA methylation/mRNA markers change with the chronological age [7]. For example, many single tissue predictors (based on methylation or expression data) have been applied to identify aging biomarkers [8]. In addition, a multi-tissue predictor based on methylation data has been used to analyze aging functions [7]. Thus predicting age using multi-scale genome-wide data in normal tissues could provide reliable results of aging-related disease risks thereby [6]. As a result, integrating multi-omics data (i.e. epigenome and transcriptome data) with high predicting ability and meaningful biological results is required to analyze the aging process.
On the other hand, finding interactions between biomarkers is also important to identify characterizations of tissue/individual changes, such as phenotype stage and disease outcome [9, 10]. Reconstructing molecular networks also gives systematic approaches to deal with multi-scale data in aging analysis [11]. A previous study has constructed cross-tissue aging networks based on tissue-specific microarray mRNA data in mouse [11]. Nevertheless, integrative aging networks from multi-scale data (i.e. methylation and expression) have not been constructed entirely in Homo species.
In the present work, we developed a computational pipeline, the Integrating and Stepwise Age-Prediction (ISAP) method, to regress and predict age by integrating methylation and expression data in 9 normal tissues of human. The improved method found key integrative markers for both multi-tissue and tissue-specific models with high accuracy. Furthermore, a serious of network analyses such as the shortest Protein-Protein Interaction (PPI) network, cross-tissue co-profiling and pathway interaction networks revealed coordinated aging patterns in both multi-omics profiling and functional levels. The results showed integrative age-correlated profiles were associated with important pathway characteristics.
Methods
Data and pre-process
We obtained paired methylation, transcription and clinical data in 9 different normal tissues with more than 10 samples (BLCA: Bladder Urothelial tissue, BRCA: Breast invasive tissue, HNSC: Head/Neck squamous cell tissue, KIRC: Kidney renal clear cell tissue, KIRP: Kidney renal papillary cell tissue, LIHC: Liver tissue, LUAD: Lung tissue, PRAD: Prostate tissue, THCA: Thyroid tissue) from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov) [12] platform (only using level-3 data). The age of each person came from TCGA clinical data. TCGA methylation data come from the Illumina Infinium Human Methylation27 BeadChip or the Illumina Infinium HumanMethylation450 BeadChip, therefore Illumina probe ID presented in both platforms were selected for further analysis [7]. Transcription data were obtained from RNASeq-v2 data (level-3). Both methylation and expression data were treated by a Singular Value Decomposition (SVD) [13] method (regress the first 3 principle components) to assess the sources of inter-sample variation separately in each tissue, and then were normalized to have zero mean and unit variance.
The choice of training data sets was guided by the following criteria the same as the previous study [7]: First, the training data should represent a wide spectrum of tissues and cell types; second, the mean age in the training data should be comparable to that of the test data. As a result, to predict age of each person in the multi-tissue model, 6 tissues (Bladder, Breast, Head/Neck, Kidney renal clear cell, Lung and Thyroid) were set as the training data (mean value ≈ 58 years). The rest 3 tissues (Kidney renal papillary cell, Liver and Prostate) were set as the independent test data (mean value ≈ 61 years). To train the multi-tissue model, each one out of the six tissues of training data was set as a set of temporary test data, so cross-validation of the multi-tissue model was performed as 6-fold. To train tissue-specific models, the common 5-fold cross-validation was performed for each tissue respectively.
Lasso regression
Least absolute shrinkage and selection operator (Lasso) is a regression method performing both variable selection and regularization to improve the prediction accuracy and interpretability of the statistical model [14]. In this work Lasso was used to regress age using methylation data and the penalty parameter λ value was determined by cross-validation.
Partial least-square (PLS) regression
The partial least-square regression (PLS) method is often used for dimension reduction when dealing with small-sized samples of gene expression data [15]. The algorithm is mainly performed as described by Höskuldsson [16].
In this work PLS was used to transformed high-dimension expression data before stepwise regression and the number of first modified direction vectors of PLS was finally determined (after stepwise regression) by cross-validation.
Stepwise regression
The stepwise regression method is also widely used to select important features in regression problem in small-sample condition [17]. In this work the method was used to select important expression features (genes), and it works in the forward style as follows:
-
(1)
Sort gene expressions by their absolute correlation coefficients with the output in descending order. In this work the output was the residuals of ages from Lasso.
-
(2)
Add each sorted gene expression after PLS transformation individually. The number of selected genes was determined by cross-validation.
Integrating and stepwise age-prediction method
In this paper, we presented an improved algorithm, the Integrating and Stepwise Age-Prediction (ISAP) method, to predict age by integrating Lasso, PLS and forward stepwise regression method based on paired methylation and expression data in normal tissues. Methylation data were used to regress age firstly, and then expression data were used to regress the residuals from methylation data. The improved computational method (ISAP) works as follows:
-
(1)
Predict age using Lasso regression based on methylation data, the penalty parameter λ value in Lasso is determined by cross-validation using training data, and save the residuals after Lasso regression.
-
(2)
Sort expression features (genes) by absolute correlation coefficients with the residuals in descending order.
-
(3)
Predict the residuals by forward stepwise regression method based on expression data. The expression data with selected features (genes) are transformed into full-ranked matrix by PLS firstly, and the number of selected genes are determined by cross-validation based on training data.
-
(4)
For selected expression features (genes), determined the number of first direction vectors of PLS by cross-validation based on the training data.
-
(5)
Caculate the final regression coefficients of aging markers (both methylation and expression), and save the regression coefficients.
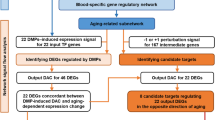
In this paper, both the multi-tissue model and tissue-specific models were calculated, and the selected aging markers were used to further functional analyses. The cross-validation was performed as 6-fold in the multi-tissue model, and 5-fold in tissue-specific models, separately. Age of both training data and test were subtracted mean value of training age before regression. The flowchart of ISAP and other succeeding works was shown in Fig. 1.

The computational pipeline (the ISAP method and further functional analyses) in this work
Enrichment analysis
To gain understandings of gene sets statistically significantly associated with biological functions, enrichment analyses were carried out. Informations of Gene Ontology (GO) terms (including all GO gene sets), GO Biological Processes (BP), and KEGG pathways were downloaded from Gene Set Enrichment Analysis (GSEA) platform (http://software.broadinstitute.org/gsea/downloads.jsp, version 5.1) [18]. The hypergeometric test [19] was performed to estimate the enrichment of these selected markers compared to known terms/pathways. The formula of the hypergeometric test is:
where N is the total gene number of the gene expression sets, M is the number of known genes sets (i.e. GO terms or KEGG pathway), n is the number of the candidate genes that we identified, and k is the number of common entries between them. P is the enrichment statistical significance of the test. Finally, the selected significantly enrichment p-values were controlled by False Discover Rate (FDR, <0.25) [18, 20].
Protein-protein interaction (PPI) network
The background weighted PPI network was constructed using data from STRING database (http://string-db.org/, version 10) [21]. It weights protein-protein interactions by calculating confidence scores. In this work, 70% confidence score (>700) has been used as a cut-off for further analysis. Each pair of selected integrative markers was picked out and calculated their shortest pathway in PPI network using Dijkstra algorithm [22]. Finally the PPI network with shortest pathway among selected markers were constructed, and proteins/genes in the PPI network were sorted by their betweennesses in descending order.
To test whether the top betweenness genes were hubs in the background network or not, we ran a permutation to count the occurrence time of the top genes in the shortest paths between random selected genes (contained the same number of selected gene set of aging markers) when they have greater betweennesses than those in our study. We repeated this process 1000 times, and the p-value was calculated as the proportion of occurrence times of the top betweenness genes in 1000 permutations.
Construction of integrative cross-tissue co-profiling network of aging
Age ≥ 60 was considered as ‘old’ age group, and age ≤ 50 was considered as ‘young’ age group. Tissues with more than 3 samples in both young and old group were selected to constructed cross-tissue network. As a result, 7 tissues were selected, they were: Breast invasive tissue, Head/Neck squamous cell tissue, Kidney renal clear cell tissue, Kidney renal papillary cell tissue, Liver tissue, Lung tissue, and Thyroid tissue. The number of tissue–tissue pairs was 21 in total.
For each tissue, profiles of selected aging markers (including both methylation and expression) were discretized using two thresholds mean+/-std. Then the discretized intertissue gene pairs (from 21 tissue-tissue pairs) within the same age group (young or old) were calculated the Kolmogorov-Smirnov (K-S) statistic [23]:
where F 1 and F 2 are the cumulative distribution of aging markers in one tissue and in another tissue, respectively. K-S values of both young age group and old age group were calculated separately. The absolute difference of K-S value between old and young group was set as the edge in the cross-tissue network, which would be filtered by a proper threshold (i.e. 0.95).
Moreover, all the aging markers in selected tissue-tissue pairs were enriched to GO terms by the hypergeometric test (FDR <0.25) to find functional characteristics of tissue-tissue cross-talk.
Construction of integrative cross-tissue pathway interaction aging network
Selected aging markers in each tissue were enriched to KEGG pathway by the hypergeometric test using formula (1). Significant KEGG pathways (FDR <0.25) in each tissue (totally 7) was selected to further analysis. Three types of pathway interaction networks were considered: first, sum of absolute K-S value differences (> a moderate threshold, i.e. 0.6) was used as the connectivity of two pathways from two tissues; second, sum of all the absolute K-S value differences (with no thresholds) was used as the connectivity of two pathways between two tissues; third, sum of all the absolute K-S value differences (with a more rigorous KEGG enrichment FDR <0.1) was used as the connectivity.
Results and discussion
Aging regression results
To identify integrative aging biomarkers of multi-tissue, Lasso regression found 164 DNA methylation markers, and then the forward stepwise regression found 77 mRNA expression markers. Totally 241 aging markers were shown in Additional file 1 as well as their regression weights. Table 1 shows that the regression results of this computational pipeline, the ISAP method, get higher accuracy (with lower residual errors) compared to other common regression methods (i.e. Lasso, elastic net or PLS, either with methylation data or expression data).
Table 1 also shows that age prediction by methylation data with higher accuracy than using expression data. Moreover, simply combining methylation and expression could not improve regression results compared with using methylation data alone (shown in Table 1). Therefore, our improved computational method could integrate methylation and transcriptional data more effectively than other general methods.
Figures 2 and 3 and Table 2 show more detailed (residual errors, Pearson correlation coefficients and median absolute difference errors) results in each tissue. The ISAP method not only got high accuracy on training tissues, but also predicted independent test tissues with enough accuracy (i.e. mean residual errors near 1.5 of each test tissue).

Regression results in the multi-tissue model. a Across all training data; b across all test data

Regression results of each tissue in multi-tissue models. a Bladder Urothelial tissue; b Breast invasive tissue; c Head/Neck squamous cell tissue; d Kidney renal clear cell tissue e Kidney renal papillary cell tissue; f Liver tissue; g Lung tissue; h Prostate tissue; i Thyroid tissue. a-d, g and i are training data; e, f and h are test data
Then we used our pipeline to regress age on each single tissue separately. Each identified integrative aging markers and their regression weights are also shown in Additional file 1. Table 3 shows regression results of tissue-specific biomarkers (details also shown in Additional file 1). Both multi-tissue and tissue-specific biomarkers were used to succeeding functional analyses (e.g. network construction).
Functional/enrichment analysis and PPI-network
Firstly, selected integrative biomarkers were sorted by their absolute regression weights in descending order, which indicated their profiling patterns correlating the chronological age. For example, the marker with the greatest weight in the multi-tissue model was methylation profile of gene GPR45. Protein of GPR45 functioned in the central nervous system, and was reported to be related to aging significantly [24]. Moreover, the marker with the greastest weight in tissue-specific models was expression of gene CORO6 in the Kidney renal clear cell model, which has been reported that to be regulated by age [25].
Furthermore, we performed enrichment analysis of selected integrative biomarkers (multi-tissue and tissue-specific) on Biological Process (BP) of Gene Ontology (GO) and KEGG pathway using the hypergeometric test. In the multi-tissue model, the top GO biological process and KEGG pathway were positive regulation of immune system process (GO:0007059, p-value = 1.6643e-08, and FDR =1.3308e-05) and cell adhesion molecules (CAMs, p-value = 1.4205e-06, and FDR =2.4517e-04), respectively. It has been reported that inflammatory gene sets such as positive regulation of immune system process or dysfunction of immune system are induced by aging [26]. In addition, multiple changes in immune system occur and disrupt the regulation of body cells with aging in the immune dysregulation theory [26], which also coincides with our results. Many cell adhesion molecules have been indicated to be dependent on aging [27], too. In tissue-specific models, the top GO biological process and KEGG pathway were negative regulation of phosphate metabolic process (GO:0045936, p-value = 9.7695e-05, and FDR =0.0157) in kidney renal papillary cell and Antigen processing and presentation pathway (p-value = 3.3052e-06, and FDR =0.0006) in thyroid, respectively. Phosphate metabolic process has been reported to relate to aging diseases and cancer [28]. Moreover, autophagy is an important mechanism of intracellular pathogen’s antigens, and dysfuncition of autophagy is also regulated by aging [29]. As a result, the enrichment analyses indicated that functions of immune system related to aging deeply.
In addition, we constructed an undirected graph with the PPI data from STRING. Each pair of selected 241 integrative markers in the multi-tissue model were picked out and calculated their shortest path by Dijkstra algorithm. Then a sub-network composed of 161 out of the 241 markers was obtained based on shortest pathways, in which a total of 44244 protein-protein interactions of 2177 proteins annotating in the Ensemble Biomart database (http://ensembl.org/biomart/martview) were picked out. These genes/proteins were ranked by their betweennesses in descending order as well as permutation p-values (shown in Table 4). There were 7 out of the top 10 betweenness genes whose p-values were significant. Among these genes, TP53 has the largest betweenness of 1023 (permutation p-value = 0.016), meaning that there are 1023 shortest paths going through this gene. Accordingly, TP53 may play an important role in connecting the 161 candidate genes and hence may be related to aging. Obviously, TP53 is one of the most important key markers in cell cycle and cell apoptosis pathway, thus the cross-talk between TP53 and MTOR regulates cellular senescence, cancer and aging [30].
Integrative aging-specific cross-tissue co-profiling networks
Aging is a complex process where many tissues and genes participate, thus gene profiling pattern pairs would be perturbed or regulated during aging. To investigate molecular aging profiling patterns of tissue-tissue pairs relating to different age group (young versus old people), cross-tissue co-profiling network were constructed using absolute difference of K-S statistics (>0.95). Finally 31 integrative inter-tissue pairs were obtained. The 31 molecular pairs belonged to 6 tissue-tissue pairs of aging, and they were shown in Fig. 4. Tissue of head/neck might be in the core status with the largest degree of 4 in the cross-tissue network, meaning that head/neck might affect other 4 tissues during the aging process.

Patterns in the tissue–tissue pairs of aging
Furthermore, each of the selected 31 pairs was calculated to investigate whether any pair shares the same GO term. As a result, the correlation of GATA4 in head/neck and EGFL7 in kidney renal clear cell shared most GO terms (number = 4), including system development (GO: 0048731), multicellular organismal development (GO: 0007275), anatomical structure development (GO: 0048856) and animal organ development (GO: 0048513). Since EGFL7 and GATA4 are related to aging [31, 32], it was possible the expression of EGFL7 in the kidney was regulated by the expression of GATA4 in head/neck, with this regulation changing with age. All the shared terms were related to cell/tissue developments, and tissue develpoments are correlated with aging [33], obviously.
In addition, all the aging specific genes with top high K-S differences (>0.85) were also performed enrichment analysis of GO terms by the hypergeometric test. The top GO terms were positive regulation of caspase activity (GO:0043280, p-value = 9.8594e-05, and FDR =0.0717). It is well known that caspase-dependent apoptotic signaling is vital to many human aging diseases [30, 34].
Integrative aging-specific cross-tissue pathway interaction networks
To find inter-tissue pathway to pathway cross-talks, cross-tissue pathway interaction aging networks were constructed. Figure 5 depicts three types of pathway interaction cross-tissue networks. The first type showed the sum of absolute differences of K-S statistic with high difference (K-S difference >0.6). Figure 5a shows the pathway interaction networks of aging among head/neck, kidney and thyroid, and interaction between cell cycle and neurotrophin signaling pathway with the largest connectivity (=2.5167) might be important to aging. The second type showed the network based on sum of absolute differences of K-S statistic (only showed sum >3). Interaction between cell adhesion molecules and neurotrophin signaling pathway was with the largest connectivity (=5.5062, shown in Fig. 5b). The third type showed sum of absolute value of K-S statistics’ difference (FDR <0.1 as the threshold), and cross-talk between kidney and thyroid was the main pattern of the network. Figure 5c shows that interaction between cell cycle and cell adhesion molecules might be critical to aging (connectivity =4.8748).

The cross-tissue pathway interaction networks of aging, and connectivities are shown in edges. a sum of absolute K-S differences (with a threshold >0.6) as connectivities; b sum of absolute K-S differences (with no thresholds) as connectivities; c sum of absolute K-S differences (FDR <0.1) as connectivities
In immunosenescence theories of aging, either innate or adaptive immune responses are related to the aging process [26]. For instance, cellular senescence is believed to be involved immune and aging progresses, and suppressed relative pathways such cell cycle, pathway of cancers and so on [26, 30, 35]. Cell adhesion cascades appear to affect the functional capacity of cells during aging [27, 36]. Moreover, both cell adhesion and neurotrophin cooperatively perform critical functions in the aging process responding to the immune system [37, 38]. Our results found that the cross-talk among key pathways (i.e. cell cycle, cell adhesion and neurotrophin signaling) played important roles in the aging process altogether. Furthermore, head/neck and kidney might be in core status to regulate the aging process and relative pathways in other tissues.
Conclusion
Predicting age in human normal tissues is fundamental to aging researches. In this paper, we developed an improved method, the ISAP pipeline, to integrate both methylation and expression data for age prediction. The ISAP method predicted age more accurately than other popular methods. Furthermore, the PPI network and enrichment analyses also find core aging genes and pathways.
In addition, network analysis could also help to identify aging related genes/pathways between different tissues. We have performed a serious of network analyses of aging specific markers, and find important profiling patterns and pathway interactions. Our results confirmed existing aging theories or hypotheses and could improve further aging researches.
References
Finch CE. Longevity, Senescence, and the Genome. Chicago: University of Chicago Press; 1990.
Xue H, Xian B, Dong D, Xia K, Zhu S, Zhang Z, et al. A modular network model of aging. Mol Syst Biol. 2007;3:147.
Gilberto L. The relationship of Parkinson disease with aging. Arch Neurol. 2007;64(9):1242–6.
McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, et al. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick’s Disease. Arch Neurol. 2001;58:1803–9.
Angela Grimes BS, Sathees BC. Chandra significance of cellular senescence in aging and cancer. Cancer Res Treat. 2009;41(4):187–95.
Sood S, Gallagher IJ, Lunnon K, Rullman E, Keohane A, Crossland H, Phillips BE, Cederholm T, Jensen T, van Loon LJ, Lannfelt L, Kraus WE, Atherton PJ, Howard R, Gustafsson T, Hodges A, Timmons JA. A novel multi-tissue RNA diagnostic of healthy ageing relates to cognitive health status. Genome Biol. 2015;16:185.
Steve H. DNA methylation age of human tissues and cell types Horvath. Genome Biol. 2013;14:R115.
Kajia Cao, Alice S. Chen-Plotkin, Joshua B. Plotkin, Li-San Wang Age-correlated gene expression in normal and neurodegenerative human brain tissues. PLoS One. 5(9): e13098. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2947518/.
Taylor IW, Linding R, Warde-Farley D, Liu Y, Pesquita C, Faria D, et al. Dynamic modularity in protein interaction networks predicts breast cancer outcome. Nat Biotechnol. 2009;27:199–204.
Huang T, Liu L, Qian Z, Tu K, Li Y, and Xie L. (2010a). Using GeneReg to construct time delay gene regulatory networks. BMC Res Notes 3, 142
Tao H, Zhang J, Lu X, Dong X, Tao H, Zhang J, Lu X, Dong X, Zhang L, Cai YD, Li YX. Crosstissue coexpression Network of aging. OMICS. 2011;15(10):665–71.
Hudson TJ, et al. International network of cancer genome projects. Nature. 2010;464:993–8.
Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Gayther SA, et al. An epigenetic signature in peripheral blood predicts active ovarian cancer. PLoS One. 2009;4:e8274.
Tibshirani R. Regression shrinkage and selection via the lasso. J Royal Stat Soc, Series B. 1996;58(1):267–88.
Boulesteix AL, Strimmer K. Partial least squares: a versatile tool for the analysis of high-dimensional genomic data. Brief Bioinform. 2006;8:32–44.
Höskuldsson A. PLS regression methods. J Chemometrics. 1988;2:211–28.
Draper N, Smith H. Applied Regression Analysis. 2nd ed. New York: Wiley; 1981.
Aravind S, Pablo T, Mootha VK, Sayan M, Ebert BL, Gillette MA, Amanda P, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. PNAS. 2005;102(43):15545–50.
Gasper G, Rahman M. Basic hypergeometric series. Cambridge: Cambridge University Press; 2004. p. xxvi–428.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J Royal Stat Soc. 1995;57:289–300.
Damian S, Andrea F, Stefan W, Kristoffer F, Davide H, Jaime H-C, Milan S, Alexander R, Alberto S, Tsafou KP, Michael K, Peer B, Jensen LJ, von Mering C. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
Dijkstra E. A note on two problems in connection with graphs. Numerische Mathematik. 1959;1:269–71.
Marsaglia G, Tsang W, Wang J. Evaluating Kolmogorov's Distribution. J Stat Software. 2003;8(18): https://www.researchgate.net/publication/5142829_Evaluating_Kolmogorov%27s_Distribution.
Park S-K, Kim K, Page GP, Allison DB, Weindruch R, Tomas A. Prolla Gene expression profiling of aging in multiple mouse strains: identification of aging biomarkers and impact of dietary antioxidants. Aging Cell. 2009;8(4):484–95.
Sanchez D, Bajo-Grañeras R, Del Caño-Espinel M, Garcia-Centeno R, Garcia-Mateo N, Pascua-Maestro R, Maria D. Ganfornina Aging without Apolipoprotein D: Molecular and cellular modifications in the hippocampus and cortex. Exp Gerontol. 2015;67:19–47.
Castelo-Branco C, Soveral I. The immune system and aging: a review. Gynecol Endocrinol. 2014;30(1):16–22.
Volker R, Fausi R, Kathrin P, Bettina H, Wolfgang R. Thomas Kuntzed Circulating Vascular Cell Adhesion Molecules VCAM-1, ICAM-1, and E-Selectin in Dependence on Aging. Gerontology. 2003;49:293–300.
Matthew D, Victor D-U, Jianhua Z. Cellular Metabolic and Autophagic Pathways: Traffic Control by Redox Signaling. Free Radic Biol Med. 2013;63:207–21.
Ana Maria C, Fernando M. Autophagy and the immune function in aging. Curr Opin Immunol. 2014;0:97–104.
Galluzzi L, Kepp O, Kroemer G. TP53 and MTOR cross-talk to regulate cellular senescence. AGING. 2010;2(9): https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2984599/.
Fitch MJ, Luisa C, Frank K, Heidi Stuhlmann E. a Novel Epidermal Growth Factor-Domain Gene Expressed in Endothelial. Cells Dev Dyn. 2004;230(2):316–24.
Chanhee K, Qikai X, Martin TD, Li MZ, Marco D, Liviu A, Tao L, Yankner BA, Judith C, Elledge SJ. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science. 2015;349(6255):aaa5612.
Richard F. Loeser Age-Related Changes in the Musculoskeletal System and the Development of Osteoarthritis. Clin Geriatr Med. 2010;26(3):371–86.
Favaloro B, Allocati N, Graziano V, Di Ilio C, De Laurenzi V. Regulation of Translation Initiation in Eukaryotes: Mechanisms and Biological Targets Cell. 2009;136(4):731–45. https://www.ncbi.nlm.nih.gov/pubmed/19239892.
Grimes A, Chandra SB. Significance of cellular senescence in aging and cancer. Cancer Res Trea. 2009;41(4):187–95.
Ponnappan S, Aging UP, Function I. Molecular Mechanisms to Interventions. Antioxid Redox Signal. 2011;14(8):1551–85. doi:10.1089/ars.2010.3228.
Quartu M, Serra MP, Boi M, Melis T, Ambu R, Del Fiacco M.Brain-derived neurotrophic factor (BDNF) and polysialylated-neural cell adhesion molecule (PSA-NCAM): codistribution in the human brainstem precerebellar nuclei from prenatal to adult age. Brain Res. 2010;1363:49-62. doi:10.1016/j.brainres.2010.09.106. Epub 2010 Oct 22.
Butcher S, Chahel H, Lord JM. Ageing and the neutrophil: no appetite for killing? Immunology. 2000;100(4):411–6.
Declarations
This article has been published as part of BMC Systems Biology Volume 10 Supplement 4, 2016: Proceedings of the 27th International Conference on Genome Informatics: systems biology. The full contents of the supplement are available online at http://bmcsystbiol.biomedcentral.com/articles/supplements/volume-10-supplement-4.
Funding
This work was supported by the National High Technology Research and Development Program (863 Program) (2012AA02A602, 2015AA020104), National Science and Technology Major Project (2012ZX09303013-015), Special funds for scientific research in the health industry (201302010).
The authors declare that publication costs of the article were funded by Natural Science Foundation of China and the National '973' Basic Research Program. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The data supporting the results of this article are included and cited within the article and its additional files.
Authors’ contributions
YW performed the algorithm and analyzed the data; YW and TH and wrote the manuscript; LL and LX designed and sponsored the study. All authors read and approved the manuscript.
Competing interests
The authors declared that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Integrative age predictors and their weights. Note: type 1 indicates methylation data, and type 0 indicates expression data; tissue with 0 indicates the multi-tissue model, and 1-9 indicate each of 9 normal tissues (Bladder Urothelial tissue, Breast invasive tissue, Head/Neck squamous cell tissue, Kidney renal clear cell tissue, Kidney renal papillary cell tissue, Liver tissue, Lung tissue, Prostate tissue, Thyroid tissue), separately. (XLS 97 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, Y., Huang, T., Xie, L. et al. Integrative analysis of methylation and transcriptional profiles to predict aging and construct aging specific cross-tissue networks. BMC Syst Biol 10 (Suppl 4), 132 (2016). https://doi.org/10.1186/s12918-016-0354-4
Published:
DOI: https://doi.org/10.1186/s12918-016-0354-4