
Abstract
Background
Pseudomonas aeruginosa is an important opportunistic pathogen in dogs and cats and is resistant to several antimicrobial drugs; however, data on the clonal distribution of P. aeruginosa in veterinary hospital are limited. This study aimed to investigate the clonal dissemination and antimicrobial resistance of clinical P. aeruginosa in a veterinary teaching hospital in Thailand within a 1-year period. Minimum inhibitory concentration determination and whole genome sequencing were used for antimicrobial susceptibility analysis and genetic determination, respectively.
Results
Forty-nine P. aeruginosa were isolated mostly from the skin, urinary tract, and ear canal of 39 dogs and 10 cats. These isolates belonged to 39 sequence types (STs) that included 9 strains of high-risk clones of ST235 (n = 2), ST244 (n = 2), ST274 (n = 2), ST277 (n = 1), ST308 (n = 1), and ST357 (n = 1). Overall antimicrobial resistance rate was low (< 25%), and no colistin-resistant strains were found. Two carbapenem-resistant strains belonging to ST235 and ST3405 were identified.
Conclusions
Clinical P. aeruginosa in dogs and cats represent STs diversity. High-risk clones and carbapenem-resistant strains are a public health concern. Nevertheless, this study was limited by a small number of isolates. Continuous monitoring is needed, particularly in large-scale settings with high numbers of P. aeruginosa, to restrict bacterial transfer from companion animal to humans in a veterinary hospital.
Similar content being viewed by others

Background
Pseudomonas aeruginosa is a ubiquitous bacterium that can survive in various environment conditions [1]. It is an important opportunistic and nosocomial pathogen in humans and is associated with cystic fibrosis, burn wound infection, ulcerative keratitis, and other infections [1, 2]. In companion animals, P. aeruginosa is commonly isolated from the skin, ears, eyes, and infected urinary tract; thus, affecting animal health and welfare [3,4,5]. Although P. aeruginosa does not often cause a life-threatening disease in companion animals, treating this pathogen is difficult when it develops the resistance to several antimicrobial agents due to various intrinsic and acquired resistance mechanisms [5, 6]. The occurrence of multidrug-resistant (MDR) and carbapenem-resistant P. aeruginosa and the co-occurrence of antimicrobial resistance genes in isolates from companion animals have been reported [4, 7, 8]. Companion animals, especially dogs and cats, could be a bacterial reservoir, and transfer P. aeruginosa or its resistance gene to humans who are in close contact and vice versa [8,9,10,11].
The clonal distribution of P. aeruginosa isolates from humans, animals, and environment is nonspecific and diverse [12]. The most important clones are those of high-risk determined through multilocus sequence typing (MLST) e.g., ST235, ST111, ST244, ST357, ST308, ST175, ST277, and ST654 [13]. These high-risk clones are distributed worldwide and can be MDR and extensively drug-resistant, which is associated with treatment failure [14]. In companion animals, a few molecular epidemiology studies of clinical P. aeruginosa were conducted [3, 4, 7, 8, 10, 15]. High-risk clones were observed in carbapenem-resistant P. aeruginosa isolates from dogs in Japan [8] and France [7]. Furthermore, our previous study identified the presence of MDR and carbapenem-resistant P. aeruginosa strains belonging to ST235, ST244, and ST606 in the environment of a veterinary teaching hospital in Thailand [16]. However, data are lacking on the sequence types and the prevalence of high-risk clones of P. aeruginosa that cause infection in dogs and cats in the veterinary hospital in Thailand.
This study aimed to investigate the sequence typing and clones, and antimicrobial resistance of clinical P. aeruginosa isolates from the dogs and cats visiting a veterinary teaching hospital in Thailand within a 1-year period. Whole genome sequencing and analysis were performed to gain a deep perspective of intrahospital clonal dissemination.
Results
Clinical characteristic of P. aeruginosa
In 2022, 1,627 samples were collected from sick dogs and cats visiting Prasu-Arthorn Veterinary Teaching Hospital, Mahidol University, Thailand. Positive bacterial cultures were prepared from 821 samples that included nonduplicate 49 P. aeruginosa (6%, 49/821). These samples were obtained from 39 dogs (79.6%, 39/49) and 10 cats (20.4%, 10/49). The most frequent isolation site was skin wound (32.7%), followed by urinary tract (28.6%) and ear canal (24.5%) (Table 1 and Additional file 1). The other sites were nasal cavity and abdominal cavity (4.1% each), as well as the oral cavity, reproductive tract, and pleural cavity (2.1% each). In dogs (n = 39), skin wound and pus on skin (35.9%) were the most frequent isolation sites. In cats, P. aeruginosa was equally isolated from the skin, ear canal, urinary tract, abdominal cavity, and nasal cavity (20%, n = 2 each) (Table 1).
Distribution of STs of clinical P. aeruginosa
P. aeruginosa belonged to 39 STs, which included 3 new STs: ST4396 (n = 1), ST4397 (n = 2), and ST4398 (n = 1) distributed in this 1-year study (Fig. 1). No predominant clones were determined from the STs; only one or two P. aeruginosa strains were included in the clones. Total of 9 strains (18.37%) belonged to the 6 high-risk clones of ST235 (n = 2), ST244 (n = 2), ST274 (n = 2), ST277 (n = 1), ST308 (n = 1), and ST357 (n = 1) (Fig. 1). These strains were isolated from various sampling sites including skin (ST235 and ST308), ear canal (ST235, ST244, and ST357), urinary tract (ST244, ST274, and ST277), and abdominal cavity (ST244) (Additional file 1). Additionally, the 6 high-risk clones were isolated during various isolation periods (Fig. 1). The isolates from the same STs were closely clustered together, according to phylogenetic SNP tree and SNP number analyses of clinical isolates from this study and 10 environmental isolates from a prior study conducted at the same veterinary hospital (Fig. 1 and Additional file 2).

Phylogenetic single nucleotide polymorphism (SNP) tree, strain, sequence types (STs), origin, isolation period, and antimicrobial resistance of Pseudomonas aeruginosa. Ten environmental isolates from a previous study [16] and the reference strain P. aeruginosa PAO1 (Accession no. AE004091.2) were included. New ST in bold. Presence in green and absence in yellow. N/A, not determined. T, cefazidime, F, cefepime, P/T, piperacillin/ tazobactam, I, imipenem, M, meropenem, A, amikacin, G, gentamicin, L, Levofloxacin, C, ciprofloxacin, and in blank, susceptible. Types of blaPDC and blaOXA; and amino acid mutation of gyrA, parE, basR, nalC in green box. oprD amino acid alterations: 1, wildtype; 2, T103S, K115T, F170L; 3, T103S, K115T, F170L, E185Q, P186G, V189T, R310E, A315G, G425A; 4, D43N, S57E, S59R, E202Q, I210A, E230K, S240T, N262T, A267S, A281G, K296Q, Q301E, R310G, V359L, 372VDSSSS–YAGL383; 5, V127L, E185Q, P186G, V189T, E202Q, I210A, E230K, S240T, N262T, T276A, A281G, K296Q, Q301E, R310E, R310E, G312R, A315G, L356M, L348M, 372VDSSSS–YAGL383, S403A, Q424E; 6, S57E, S59R, V127L, E185Q, P186G, V189T, E202Q, I210A, E230K, S240T, N262T, T276A, A281G, K296Q, Q301E, R310E, A315G, L347M, 372VDSSSS–YAGL383, S413A, Q424E
Antimicrobial resistance of P. aeruginosa
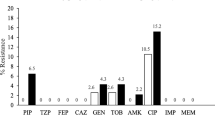
Among the P. aeruginosa isolates, 16 were resistant to at least one of the drugs tested and 33 (67.4%) were susceptible strains. For the 16 strains, the highest phenotypic resistant rate was observed for gentamicin (24.5%), followed by amikacin (14.3%), ciprofloxacin (14.3%), and levofloxacin (12.2%). Resistance to piperacillin/ tazobactam and imipenem accounted for 4.1% each, and resistance to ceftazidime, cefepime, and meropenem was at 2%. No colistin resistance was observed. Two MDR strains (1184 and 143) were identified (2%, 2/49) and found to be carbapenem-resistant strains. Strain 143 was resistant to imipenem, gentamicin, and ciprofloxacin; and strain 1184 was resistant to ceftazidime, cefepime, piperacillin/tazobactam, imipenem, meropenem, gentamicin, amikacin, levofloxacin, and ciprofloxacin (Fig. 1).
Genes responsible for acquired and phenotypic antimicrobial resistance and those with amino acid alterations were selected from the Comprehensive Antibiotic Resistance Database (CARD) database for analysis in Fig. 1. In general, the strains belonging to the same STs exhibited a similar antimicrobial resistance gene profile, except for ST235 and ST244. All of them harbored different types of β-lactamases, blaPDC and blaOXA (Fig. 1). The combination of blaVIM−2, blaPDC−35 and blaOXA−488 was detected in a single strain 1184-ST235 that was resistant to imipenem and meropenem. While two carbapenem-resistant ST235 isolates from the environment (strains WA1.3 and WA1.1) contained blaVEB−1, blaPDC−35, blaOXA−488, and blaOXA−10 (Fig. 1). Aminoglycoside resistance gene aph(3’)-Ilb was detected in all the isolates alone or in combination with the others. The gentamicin and/or amikacin resistance genes were aph(3’’)-Ib, aph(6)-Id in ST308, ant(2’’)-Ia, ant(3‘’)-Ila, ant(4’)-Ilb, aadA6, and aadA2 in ST235; aph(3’)-Vla in ST620; and aac(6’)-Ib9 and aadA2 in ST3405. A single strain ST244 harboring aac(6’)-Ib8 and ant(3‘’)-Ila was susceptible to gentamicin and amikacin. Except for aph(3’)-Ilb, none of the aminoglycoside resistance genes were found in the seven gentamicin- and/or amikacin-resistant strains. The mutation in gyrA (T83I) but not in parE (A473V) or qnrVC1 was associated with resistance to ciprofloxacin and levofloxacin in clinical isolates. The disinfectant resistance gene, qacEdelta1, was identified in two strains of ST235 and a single strain 547-ST3405. A sulfonamide resistance gene (sul1) and a tetracycline resistance gene [tet(C)] were found in strain 240-ST308, and trimethoprims (dfrA14) and tet(A) were found in strain 39-ST244. Similar to strain 1184-ST235, strain 39-ST244 also contained the chloramphenicol resistance genes, cmlA6 and cmlA9. A point mutation of basR (L71R) gene associated with polymyxcin B resistance was observed at 75.5% (n = 37). None of nalC hit search (n = 2), wild type (n = 2), and mutation in the nalC gene of efflux pump regulatory genes at G71E (n = 11), A186T/G71E (n = 5), and S209R and G71E (n = 29) were found. Among the 49 strains, 45 had several polymorphisms of the amino acid alterations of oprD (Fig. 1).
Discussion
The several STs with genetic diversity of clinical P. aeruginosa disseminated in a veterinary teaching hospital were discovered by MLST and whole genome analysis. Clinical P. aeruginosa was mainly isolated from the skin, urinary tract, and ear canal, which in consistent with a previous study but shows a different prevalence [17, 18]. The genomic diversity of P. aeruginosa was observed from different infection types (keratitis and cystic fibrosis) in humans [19]. However, no specific clone was found on each sampling or infection sites during MLST and phylogenetic SNP tree analysis in the present work.
Six high-risk clones of ST235, ST244, ST274, ST277, ST308, and ST357 circulated in the dogs and cats in this study. The dissemination of ST235 and ST244 was previously reported in the environment of the same veterinary hospital in 2020 [16]. ST244 is a frequently found clone but is not necessarily associate with antimicrobial resistance [13], which supports the findings in this recent study. Among them, ST235 is the most relevant and widespread high-risk clone worldwide; it produces different carbapenemases in association with horizontally acquired resistance and harbors high virulence due to the production of the potent type III secretion system exotoxin encoded by exoU gene [13]. In addition, carbapenem-resistant P. aeruginosa ST235 was the predominant clone in the clinical isolates of dogs and cats in Japan [8]. This recent study found that strain 1184-ST235 was resistant to β-lactams, carbapenem, aminoglycosides, and fluoroquinolone. Resistance to imipenem and meropenem in strain 1184-ST235 mediated by the VIM-2 metallo-β-lactamase (blaVIM−2) of P. aeruginosa is the most common metallo-β-lactamase gene in many regions of Thailand and Asia [4, 20, 21]. Meanwhile, ST235 isolates from the environment of the same veterinary hospital contained blaVEB−1 [16]. Therefore, ST235 has high potential to be distributed in the dogs and cats and environment of the veterinary hospital with different antimicrobial resistance levels.
Apart from the blaVIM−2 gene, several types of β-lactamase genes blaPDC and blaOXA were identified in all the isolates. PDC and OXA types are the predominant chromosomal β-lactamase in P. aeruginosa [22]. The different combinations of aph(3’’)-Ib, aph(6)-Id, aadA2, aadA6, qacEdelta1, dfrA14, sul1, cmlA6, and cmlA9 could represent the integron class I [14, 19] in strains 240-ST308, 1056-ST235, 1184-ST235, and 547-ST3405. Integron class I with diverse gene cassette array is common in P. aeruginosa, especially in the high-risk clones, and is associated with horizontally acquired antimicrobial resistance gene and MDR [14, 20].
The overall resistance rate in this study was low (< 25%). The resistance rate of gentamicin (24.5%) was the highest rate, which was also discovered in a previous investigation of P. aeruginosa isolate from dogs in Korea (26.3%) [4] and the isolates from the environment in the same veterinary hospital (47.4%). Gentamicin is frequently used as a topical drug to treat otitis externa and skin infection in this veterinary hospital, and this practice could influence its resistance. The resistance rate to carbapenem (4.1%), including imipenem (4.1%) and meropenem (2%), was lower in the current work than that in dogs in Korea (imipenem 12.5% and meropenem 16.3%) [4], animals in France (5.7%) [7], dogs and cats across Japan (imipenem 6.7% and meropenem 2.1%) [8]. Carbapenem is preserved and restrict to treat severe infection in the veterinary teaching hospital in the present study. Although the carbapenem resistance rate was low, the increasing prevalence of carbapenem-resistant P. aeruginosa has been reported in other monitoring studies [7, 8]. Therefore, the continuous monitoring of MDR and carbapenem-resistant is warranted, especially in the veterinary hospital in many regions of Thailand. With regard to the absence of genes for the phenotypic resistance of a single imipenem-resistant strain and seven amikacin- and/ or gentamicin-resistant strains, other mechanisms, including the overexpression of efflux pumps e.g., MexXY and MexEF-OprN, and the accumulation of multiple mutations, may be responsible for this antimicrobial resistance [6, 14].
Conclusion
Diverse STs of clinical P. aeruginosa were disseminated in a veterinary teaching hospital in Thailan during a 1-year period. Six high-risk clones of ST235, ST244, ST274, ST277, ST308, and ST357 were observed without predominance and specificity to the sampling sites. This study revealed the low antimicrobial resistance rate, including carbapenem resistance and MDR. The limitation of this study included the small number of isolates from a single veterinary hospital. The expanded scale of monitoring and surveillance in more than one veterinary hospital is necessary to assess the situation and design the prevention strategy for veterinary medicine.
Methods
Bacterial isolates
P. aeruginosa was isolated from various infection sites of sick dogs and cats visiting Prasu-Arthorn Veterinary Teaching Hospital, Mahidol University, Thailand during January–December 2022. The samples were collected by the veterinarians and were sent for routine species identification at the Veterinary Diagnostic Center of the Faculty of Veterinary Science, Mahidol University, Thailand. The samples were cultured on tryptic soy agar (TSA) with 5% sheep blood (Biomedia, Thailand) and MacConkey agar (Biomedia, Thailand), and were incubated at 35 ± 2 °C for 24 h. Single colony was subcultured on TSA with 5% sheep blood agar and incubated at 35 ± 2 °C for 24 h in order to obtain homogeneous colonies. The species identification was performed using biochemical tests, including oxidase test, citrate test, motility tests, triple sugar iron reaction, and production of yellow-green or blue-green diffusible pigment on King A and King B media. The isolates identified as P. aeruginosa were included in this study. Their species were confirmed by MALDI-TOF MS (Bruker Daltonics, Germany).
Antimicrobial susceptibility testing
The minimum inhibitory concentration (MIC) for 10 antipseudomonal drugs was determined using THN1F Sensititre broth microdilution following the manufacturer’s recommendations (Sensititre, Thermo Scientific, Waltham, MA, USA). The breakpoints for ceftazidime, piperacillin/tazobactam, imipenem, levofloxacin, amikacin, and gentamicin were interpreted following the guidelines available for animals [23], and the breakpoints used for cefepime, tetracycline, meropenem, ciprofloxacin, and colistin were interpreted following the breakpoints for humans [24]. The P. aeruginosa isolates were grown on 5% sheep blood agar and incubated at 37 °C for 24 h before use. Escherichia coli ATCC 25922 was used as positive control for MIC determination. MDR was defined as the isolate being resistant to at least one agent in ≥ 3 classes of the antimicrobial drugs tested [25]. The intermediate interpretation was considered as susceptible following the definition of susceptibility testing categories by EUCAST (www.eucast.org).
Whole genome sequencing and analysis
Genomic DNA was extracted using commercial kits (ZymoBiomics DNA miniprep kit, ZymoResearch, USA) following the manufacturer’s instructions. The genomes were sequenced using Illumina NovaSeq 6000 (150 bp paired end, Illumina, San Diego, USA), and the raw reads were processed using fastp v.0.23.3 [26]. De novo assembly was performed using SPAdes in Shovill v.1.1.0 [27, 28]. The contigs size of ≥ 200 bp were used for gene annotation in Prokka v.1.14.6 [29]. The antimicrobial resistant genes were identified using the CARD [30]. The sequence of the oprD gene was compared with the reference strain P. aeruginosa PAO1 (Accession no. AE004091.2) using Clustal Omega of EMBL-EBI website (https://www.ebi.ac.uk/Tools/msa/clustalo/). MLST was analyzed for sequence types (STs) in pubMLST website (https://pubmlst.org) [31]. The phylogenetic tree of P. aeruginosa was constructed from the concatenated sequence of core-single nucleotide polymorphisms (core-SNPs) located in the region of core-genome of 49 P. aeruginosa from this study, with P. aeruginosa strain PAO1 as the reference strain (accession no. AE004091), and 10 environmental isolates from the previous study [16]. The core sequences were aligned by Parsnp program with the default parameter settings [32], and the recombination regions were filtered off using Gubbins [33]. The SNP sites were then called and concatenated by snippy (https://bactopia.github.io/bactopia-tools/snippy/). The phylogenetic tree was constructed using the maximum likelihood method with the GTR + Γ substitution model and 1,000 bootstrap replicates in the IQTREE 2 program [34]. The tree was visualized with Itol v6 online version (https://itol.embl.de/) [35].
Data Availability
This whole genome project has been deposited at GenBank of NCBI under bioproject PRJNA1001300. The accession are JAUZAE000000000- JAUZAZ000000000, JAUZBA000000000-JAUZBZ000000000, and JAVCNP000000000. The genomic data of this article is available in GenBank of NCBI repository, https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1001300. The datasets supporting the conclusions of this article are included within the article and its additional file 1 and additional file 2.
Abbreviations
- P. aeruginosa:
-
Pseudomonas aeruginosa
- MLST:
-
Multilocus sequence typing
- ST:
-
Sequence typing
- VIM-2:
-
Verona imipenemase-2
- MALDI-TOF:
-
MS Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometer
- CLSI:
-
Clinical & Laboratory Standards Institute
- EUCAST:
-
European Committee on Antimicrobial Susceptibility Testing
- ATCC:
-
American Type Culture Collection
- SNP:
-
Single nucleotide polymorphism
- CARD:
-
Comprehensive Antibiotic Resistance Database
References
Wu W, Jin Y, Bai F, Jin S. Chap. 41-Pseudomonas aeruginosa. In: Tang TW, Sussman M, Liu D, Poxton I, Schwartzman J, editors. Molecular Medical Microbiology. 2nd ed. Cambridge, MA, USA: Academic; 2015. pp. 753–67.
Patil S, Chen X, Dong S, Mai H, Lopes BS, Liu S, et al. Resistance genomics and molecular epidemiology of high-risk clones of ESBL-producing Pseudomonas aeruginosa in young children. Front Cell Infect Microbiol. 2023;13:1168096.
Lin D, Foley SL, Qi Y, Han J, Ji C, Li R, et al. Characterization of antimicrobial resistance of Pseudomonas aeruginosa isolated from canine infections. J Appl Microbiol. 2012;113(1):16–23.
Hyun JE, Chung TH, Hwang CY. Identification of VIM-2 metallo-β-lactamase-producing Pseudomonas aeruginosa isolated from dogs with pyoderma and otitis in Korea. Vet Dermatol. 2018;29(3):186–e68.
EFSA AHAW Panel (EFSA Panel on Animal Health and Welfare), Nielsen SS, Bicout DJ, Calistri P, Canali E, et al. Assessment of listing and categorisation of animal diseases within the framework of the Animal Health Law (Regulation (EU) 2016/429): antimicrobial-resistant Pseudomonas aeruginosa in dogs and cats. EFSA J. 2022;20(5):e07310.
Lopez-Causape C, Cabot G, Del Barrio-Tofino E, Oliver A. The versatile mutational resistome of Pseudomonas aeruginosa. Front Microbiol. 2018;9:685.
Haenni M, Bour M, Chatre P, Madec JY, Plesiat P, Jeannot K. Resistance of animal strains of Pseudomonas aeruginosa to carbapenems. Front Microbiol. 2017;8:1847.
Hayashi W, Izumi K, Yoshida S, Takizawa S, Sakaguchi K, Iyori K, et al. Antimicrobial resistance and type III secretion system virulotypes of Pseudomonas aeruginosa isolates from dogs and cats in primary veterinary hospitals in Japan: identification of the international high-risk clone sequence type 235. Microbiol Spectr. 2021;9(2):e00408–21.
Mohan K, Fothergill JL, Storrar J, Ledson MJ, Winstanley C, Walshaw MJ. Transmission of Pseudomonas aeruginosa epidemic strain from a patient with cystic fibrosis to a pet cat. Thorax. 2008;63(9):839–40.
Haenni M, Hocquet D, Ponsin C, Cholley P, Guyeux C, Madec JY, et al. Population structure and antimicrobial susceptibility of Pseudomonas aeruginosa from animal infections in France. BMC Vet Res. 2015;11:9.
Fernandes MR, Sellera FP, Moura Q, Carvalho MPN, Rosato PN, Cerdeira L, et al. Zooanthroponotic transmission of drug-resistant Pseudomonas aeruginosa. Brazil Emerg Infect Dis. 2018;24(6):1160–2.
Kidd TJ, Ritchie SR, Ramsay KA, Grimwood K, Bell SC, Rainey PB. Pseudomonas aeruginosa exhibits frequent recombination, but only a limited association between genotype and ecological setting. PLoS ONE. 2012;7(9):e44199.
Del Barrio-Tofino E, Lopez-Causape C, Oliver A. Pseudomonas aeruginosa epidemic high-risk clones and their association with horizontally-acquired β-lactamases: 2020 update. Int J Antimicrob Agents. 2020;56(6):106196.
Oliver A, Mulet X, Lopez-Causape C, Juan C. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist Updat. 2015;21–22:41–59.
Morris DO, Davis MF, Palmeiro BS, O’Shea K, Rankin SC. Molecular and epidemiological characterization of canine Pseudomonas otitis using a prospective case-control study design. Vet Dermatol. 2017;28(1):118–e25.
Soonthornsit J, Pimwaraluck K, Kongmuang N, Pratya P, Phumthanakorn N. Molecular epidemiology of antimicrobial-resistant Pseudomonas aeruginosa in a veterinary teaching hospital environment. Vet Res Commun. 2023;47(1):73–86.
Harada K, Arima S, Niina A, Kataoka Y, Takahashi T. Characterization of Pseudomonas aeruginosa isolates from dogs and cats in Japan: current status of antimicrobial resistance and prevailing resistance mechanisms. Microbiol Immunol. 2012;56(2):123–7.
Yukawa S, Tsuyuki Y, Sato T, Fukuda A, Usui M, Tamura Y. Antimicrobial resistance of Pseudomonas aeruginosa isolated from dogs and cats in primary veterinary hospitals in Japan. Jpn J Infect Dis. 2017;70(4):461–3.
Subedi D, Vijay AK, Kohli GS, Rice SA, Willcox M. Comparative genomics of clinical strains of Pseudomonas aeruginosa strains isolated from different geographic sites. Sci Rep. 2018;8(1):15668.
Kim MJ, Bae IK, Jeong SH, Kim SH, Song JH, Choi JY, et al. Dissemination of metallo- β-lactamase-producing Pseudomonas aeruginosa of sequence type 235 in Asian countries. J Antimicrob Chemother. 2013;68(12):2820–4.
Khuntayaporn P, Yamprayoonswat W, Yasawong M, Chomnawang MT. Dissemination of carbapenem-resistance among multidrug resistant Pseudomonas aeruginosa carrying metallo- β-lactamase genes, including the novel bla(IMP-65) gene in Thailand. Infect Chemother. 2019;51(2):107–18.
Zhao WH, Hu ZQ. β-lactamases identified in clinical isolates of Pseudomonas aeruginosa. Crit Rev Microbiol. 2010;36(3):245–58.
CLSI VET01S. Performance Standards for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated From Animals. 6th ed. Wayne, PA. 2023.
CLSI M100. Performance Standards for Antimicrobial Susceptibility Testing. 33rd ed. Wayne, PA. 2023.
Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect. 2012;18(3):268–81.
Chen S, Zhou Y, Chen Y, Gu J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34(17):i884–90.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19(5):455–77.
Seemann T, Shovill. Faster SPAdes assembly of Illumina reads 2017. https://github.com/tseemann/shovill.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30(14):2068–9.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–73.
Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018;3:124.
Todd J, Treangen BDO, Sergey Koren, Adam M, Phillippy. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15(524).
Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43(3):e15.
Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37(5):1530–4.
Letunic I, Bork P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49(W1):W293–6.
Acknowledgements
We thank the Veterinary Diagnostic Center of the Faculty of Veterinary Science, Mahidol University for providing the isolates and data.
Funding
Open access funding provided by Mahidol University. This research paper is supported by Specific League Funds from Mahidol University.
Open access funding provided by Mahidol University
Author information
Authors and Affiliations
Contributions
NP designed, funded, and coordinate this study. AJ and SA performed the experiment. NP and KL worked with bioinformatics analysis. NP wrote the first draft manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The use of isolates collected from animals in this project was approved by the Faculty of Veterinary Science, Mahidol University-Institute Animal Care and Use Committee (FVS-MU-IACUC) (MUVS-2023-04-25). We stated to confirm that all methods were carried out in accordance with relevant guidelines and regulations.
Competing interests
The authors declare that they have no competing interest
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jangsangthong, A., Lugsomya, K., Apiratwarrasakul, S. et al. Distribution of sequence types and antimicrobial resistance of clinical Pseudomonas aeruginosa isolates from dogs and cats visiting a veterinary teaching hospital in Thailand. BMC Vet Res 20, 234 (2024). https://doi.org/10.1186/s12917-024-04098-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-024-04098-5