
Abstract
Background
Feline herpesvirus type 1 (FHV) and Feline calicivirus (FCV) are the primary co-infecting pathogens that cause upper respiratory tract disease in cats. However, there are currently no visual detection assays available for on-site testing. Here, we develop an ultrasensitive and visual detection method based on dual recombinase polymerase amplification (dRPA) reaction and the hybrid Cas12a/Cas13a trans-cleavage activities in a one-tube reaction system, referred to as one-tube dRPA-Cas12a/Cas13a assay.
Results
The recombinant plasmid DNAs, crRNAs, and RPA oligonucleotides targeting the FCV ORF1 gene and FHV-1 TK gene were meticulously prepared. Subsequently, dual RPA reactions were performed followed by screening of essential reaction components for hybrid CRISPR-Cas12a (targeting the FHV-1 TK gene) and CRISPR-Cas13a (targeting the FCV ORF1 gene) trans-cleavage reaction. As a result, we successfully established an ultra-sensitive and visually detectable method for simultaneous detection of FCV and FHV-1 nucleic acids using dRPA and CRISPR/Cas-powered technology in one-tube reaction system. Visual readouts were displayed using either a fluorescence detector (Fluor-based assay) or lateral flow dipsticks (LDF-based assay). As expected, this optimized assay exhibited high specificity towards only FHV-1 and FCV without cross-reactivity with other feline pathogens while achieving accurate detection for both targets with limit of detection at 2.4 × 10− 1 copies/μL for the FHV-1 TK gene and 5.5 copies/μL for the FCV ORF1 gene, respectively. Furthermore, field detection was conducted using the dRPA-Cas12a/Cas13a assay and the reference real-time PCR methods for 56 clinical samples collected from cats with URTD. Comparatively, the results of Fluor-based assay were in exceptional concordance with the reference real-time PCR methods, resulting in high sensitivity (100% for both FHV-1 and FCV), specificity (100% for both FHV-1 and FCV), as well as consistency (Kappa values were 1.00 for FHV-1 and FCV). However, several discordant results for FHV-1 detection were observed by LDF-based assay, which suggests its prudent use and interpretaion for clinical detection. In spite of this, incorporating dRPA-Cas12a/Cas13a assay and visual readouts will facilitate rapid and accurate detection of FHV-1 and FCV in resource-limited settings.
Conclusions
The one-tube dRPA-Cas12a/Cas13a assay enables simultaneously ultrasensitive and visual detection of FHV-1 and FCV with user-friendly modality, providing unparalleled convenience for FHV-1 and FCV co-infection surveillance and decision-making of URTD management.
Similar content being viewed by others

Background
Upper respiratory tract disease (URTD) is a prevalent condition that affects cats of all ages [1]. Feline patients exhibit symptoms like sneezing, conjunctivitis, and nasal or ocular discharge, ranging from mild to severe illnesses, which is commonly caused by viral or bacterial agents, such as feline herpesvirus type-1 (FHV-1), feline calicivirus (FCV), Mycoplasma felis, Chlamydia felis, and Bordetella bronchiseptica [1, 2]. Additionally, several studies also sporadically documented the relationships between URTD and feline panleukopenia virus (FPV), feline coronavirus (FCoV), Escherichia coli or Klebsiella pneumoniae, etc. [2,3,4,5]. Increasing epidemiological investigations have revealed that co-infection of FHV-1 (the order of α-Herpesvirinae and the family of Herpesviridae, DNA virus) and FCV (the order of Picornavirales and the family of Caliciviridae, RNA virus) accounts for over 20 ∼ 50% of URTD cases in multi-cat household or high-stress environments [6, 7], potentially leading to reactivation of FCV or FHV-1 in feline carriers and reinfection [8, 9]. Moreover, the reduced virus shedding in mildly infected cats or asymptomatic individuals challenge to the sensitivity and efficiency of current molecular diagnostics targeting FHV-1 and FCV in veterinary practices. Therefore, there is an urgent need for the development of an ultrasensitive diagnostic approach suitable for on-site detection and early surveillance of FHV-1 and FCV infection.
Various real-time PCR methods for detecting FHV-1 or FCV have been well developed [6, 10,11,12,13], but their clinical applications is limited due to the requirement for well-trained personnel, PCR amplification using a thermal cycler, and time-consuming processes especially for real-time detection. Therefore, novel methodologies for rapid nucleic acid detection based on isothermal amplification technology, such as recombinase polymerase amplification (RPA) or its variant [14, 15], have emerged as robust alternatives for the rapid detection of FHV-1 or FCV nucleic acids. However, the potential non-specific amplification may compromise the specificity of these detection methods. To address this concern, CRISPR-based (Clustered regularly interspaced palindromic repeats) biosensors coupled with RPA have been employed to detect epidemic FHV-1 or FCV variants [16, 17], which have demonstrated higher sensitivity compared to the real-time PCR technology [16, 18] and other isothermal amplification approaches [14, 15]. In the integrated RPA/CRISPR-Cas reaction system, the designed guide RNA (gRNA) will specifically recognize target gene sequence (RPA products), thereby activating the trans-cleavage activity of CRISPR-Cas nucleases to generate detectable signals by cleaving ssDNA or ssRNA reporters [16, 17]. Nevertheless, simultaneous detection of FHV-1 and FCV nucleic acid has not yet been achieved in a one-tube CRISPR-powered reaction system.
CRISPR-Cas12a and CRISPR-Cas13a orthologs, such as LbaCas12a and LwaCas13a, derived from diverse bacterial species, are commonly utilized for molecular detection. Accordingly, the Cas12a-based DETECTR (DNA Endonuclease-Targeted CRISPR Trans Reporter) [19] and Cas13a-based SHERLOCK (Specific High Sensitivity Enzymatic Reporter Unlocking) platforms [20] have been developed and extensively employed as diagnostic tools. In a previous study, the cost-effective and efficient approach was developed by exploiting Cas13 homologs (Cas13a and Cas13b) and Cas12a to enable multiplexed viruses discrimination in a single system [21]. However, complete elimination of fluorescence interference in multiple Cas13-powered system remains an ongoing challenge, which may possibly result in ambiguous interpretation when using fluorescence or colorimetric visual readouts. In another study, a lateral flow assay incorporating CRISPR/Cas9 with multiplex RT-RPA was established to simultaneously detect the E and Orf1ab genes of SARS-CoV-2 while reduced detection capability for suspected clinical samples (cycle threshold values over 36) was observed [22]. Recently, LbCas12a and LbuCas13a were assembled to detect the N and Orf1ab genes of SARS-CoV-2 using a handheld fluorescence detector [23], which generated specific fluorescence readouts without signal interference. Nevertheless, the detection efficiency for dual genes in several clinical samples was not consistent compared to the RT-qPCR methods [23], suggesting the need for further improvement to achieve balanced and saturated fluorescence signals for multiple targets detection. Additionally, a hybrid CRISPR-Cas12a/Cas13a system has also been successfully developed for duplex detection of SARS-CoV variants (N and Orf1ab gene), which was combined with multiplex lateral flow assay. [24]. This innovative approach provided adevice-independent alternative for multiple genes detection. Moreover, similar hybrid CRISPR-Cas system has been successfully applied to detect crop pathogens (cauliflower mosaic virus and rhizobium radiobacter), human papillomaviruses (HPVs), or exosomal proteins coupling with multiplex-PCR or multiplex-RPA assays [25,26,27,28]. Although these findings highlight the flexibility of hybrid CRISPR-Cas system adapted for various targets detection. Further efforts are needed to enhance the efficiency for detecting the targets of interest regarding the kinetic equilibrium of molecular determinants involved in the hybrid reaction system.
In this study, we developed a novel approach, referred to as the dRPA-Cas12a/Cas13a assay, by integrating dual RPA (dRPA) reactions with a hybrid Cas12a/Cas13a trans-cleavage system for simultaneous detection of FHV-1 and FCV nucleic acids, and validated its detection efficacy in clinical settings.
Results
Dual RPA reaction
To assess the efficacy of the RPA reaction, purified products obtained from single RPA and dual RPA were submitted for 2% gel electrophoresis analysis. The dual RPA reaction yielded two distinct products as single RPA reaction did with high efficiency (Supplementary Fig. 1), which provided both FHV-1 TK gene and FCV ORF1 gene templates for subsequent utilization in one-tube CRISPR-Cas12a/Cas13a trans-cleavage system.
Visual Readouts of one-tube dRPA-Cas12a/Cas13a assay
To assess the performance of the one-tube dRPA-Cas12a/Cas13a assay, the detection results were visualized using a multi-color fluorescence detector (Fluor-based assay) and lateral flow dipstick (LDF-based assay), respectively. As the Cas12a and Cas13a trans-cleavage activities were efficiently triggered by specific crRNA-target recognition, detectable signals in different exciting light channels or lateral flow dipsticks were generated. It was expected that detection signals of FCV ORF1 gene and FHV-1 TK gene were observed in single channel or double channel within the one-tube reaction systems, demonstrating no mutual interference between Cas12a and Cas13a-based reactions (Fig. 1). Notably, reduced fluorescence intensity occurred in both fluorescence channels in the preliminary one-tube reaction system.

Preliminary establishment of dRPA-Cas12a/Cas13a assay. Positive detection readouts for single FHV-1 gene, single FCV gene, and double targets were obtained using preliminary dRPA-Cas12a/Cas13a assay without signal disturbance by fluorescence (A) and lateral flow dipstick modality (C), while unoptimized duplex reaction system resulted in reduced fluorescence intensity for both targets compared to initial single target (B). NC: negative control (DNase/RNase-free water). The statistical significance indicated by asterisks, ***P < 0.001
Optimization of one-tube dRPA-Cas12a/Cas13a assay
The one-tube dRPA-Cas12a/Cas13a assay successfully achieved simultaneous detection of dual targets in the preliminary reaction system, but reduced detection capabilities for both targets were still observed. Therefore, we optimized several crucial components of one-tube reaction systems to obtain a maximum detection capability equivalent to the corresponding single assay. As shown in the results, increasing amounts of dual RPA products enhanced fluorescence intensity and resulted in balanced fluorescence signals in serial screenings. Among them, 4 μL of dual RPA products were determined as the optimal and cost-efficient amount in the 30 μL one-tube reaction systems (Supplementary Fig. 2). To investigate whether competitive inhibition between Cas12a and Cas13a activities would occur and subsequently impair the detection capabilities for both targets, we tailored the amounts of RNase inhibitor and T7 RNA polymerase mix (the exclusive components for Cas13a-based assay). As expected, increasing amounts of RNase inhibitor enhanced Cas13a activity while slightly inhibiting Cas12a activity, while additional amounts of RNase inhibitor (over 24 U) didn’t have remarkable effect on both fluorescence signals (Supplementary Fig. 3). Furthermore, gradual increments of T7 RNA polymerase mix (25 ∼ 100 U) could promote the Cas13a activity. However, its further accumulation (125 ∼ 150 U) in the one-tube reaction systems significantly compromised the Cas12a activity (Supplementary Fig. 4). Finally, the optimal amounts for RNase inhibitor and T7 RNA polymerase mix were determined as 24U and 100 U, respectively, in current one-tube dRPA-Cas12a/Cas13a assay.
Specificity and sensitivity of one-tube dRPA-Cas12a/Cas13a assay
The specificity and sensitivity of the optimized one-tube assay were further tested, respectively. As a result, the optimized one-tube assay exclusively detected nucleic acids of FHV-1 and FCV, exhibiting no cross-reactivity with other feline pathogens. (M. felis, C. felis, B. bronchiseptica, FCoV, FPV, K. peumoniae and E. coli) (Fig. 2). The initial concentration of FHV-1 and FCV plasmid DNA were previously determined as 2.35 × 107 copies/μL and 5.5 × 107 copies/μL, respectively. In serial dilution testings, the limit of detection (LOD) for FHV-1 and FCV were 2.4 × 10− 1 copies/μL and 5.5 copies/μL, respectively (Fig. 3).

Specificity of dRPA-Cas12a/Cas13a assay. (A) The FHV-1, FCV, and feline-associated pathogens were detected using optimized dRPA-Cas12a/Cas13a assay. This assay only detected FHV-1 and FCV simultaneously by fluorescence (A) and lateral flow dipstick (C) modality, and no cross-reaction with other feline pathogens was found. (B) The balanced fluorescence signals for FHV-1 and FCV was observed in the one-tube reaction systems. NC: negative control (DNase/RNase-free water)

Sensitivity of dRPA-Cas12a/Cas13a assay. Serial dilutions (107-10− 2) of FHV-1 and FCV plasmid DNA were tested using optimized dRPA-Cas12a/Cas13a assay. The limit of detection (LOD) for FHV-1 and FCV were at 10− 1 and 100 dilution, respectively, by fluorescence (A) and lateral flow dipstick (C) modality. (B) The detection capabilities for FHV-1 and FCV were nearly parallel in serial dilutions, while the LOD for FHV-1 (10-1 dilution) was lower than FCV (100 dilution). The statistical significance was indicated by asterisks, ***P < 0.001. NC: negative control (DNase/RNase-free water)
Clinical detection and concordance with reference real-time PCR methods
The clinical utility of one-tube dRPA-Cas12a/Cas13a assay was verified by testing 56 swab samples and comparing the detection results with that of reference real-time PCR methods (FHV-1 qPCR and FCV RT-qPCR). The results of Fluor-based assay and LDF-based assay were in agreement for FCV detection. However, the discordant results of S4, S9, S28, S35 and S44 were observed for FHV-1 detection (Supplementary Figures S5-S7). Based on the Ct values by the reference real-time PCR methods, the Fluor-based assay was considered as a reliable visual modality for FCV and FHV-1 detection. When referring to the default cut-off Ct value (below 36), the positive detection rates of FCV using dRPA-Cas12a/Cas13a-Fluor assay and the reference real-time PCR were 37.5% (95% CI: 24.4–50.6%) and 23.2% (95% CI: 11.8–34.6%), respectively. Similarly, the detection rates for FHV-1 were 33.9% (95% CI: 21.1–46.7%) and 23.2% (95% CI: 11.8–34.6%) using these two methods, respectively, as shown in Fig. 4. Comparatively, the concordance for detection of positive samples (Ct value below 36) between dRPA-Cas12a/Cas13a-Fluor assay and the reference real-time PCR methods was observed. In addition, Sanger sequencing also identified several suspected positive samples (Ct values between 36 and 38) as true positives for FHV-1 (S15, S16, S21, S29, S45, S52) or FCV (S7, S15, S25, S31, S37, S42, S49, S54), as shown in Supplementary Fig. 6. This finding substantially improved the consistency for FHV-1 and FCV detection between these two methods, and generated a portion of FHV-1/FCV positive (12.5%, n = 7/56). As a result, the dRPA-Cas12a/Cas13a-Fluor assay demonstrated a sensitivity of 100%, specificity of 100%, and kappa value of 1.00 for FHV-1 and FCV detection (Table l), indicating its exceptional performance in a clinical setting.

Heat-map of detection results for 56 clinical samples. Current one-tube dRPA-Cas12a/Cas13a-Fluor assay yielded more FHV-1 and FCV positive results compared to the reference real-time PCR methods, when the cut-off Ct value was set as below 36. Additionally, the suspected positive samples yielded by the real-time PCR methods (Ct value was between 36 and 38) were marked by blue stellar. NC: negative control (DNase/RNase-free water)
Discussion
In present study, we developed a novel duplex diagnostic method, namely the one-tube dRPA-Cas12a/Cas13a assay, which employed a visual modality for detection of emerging FHV-1 and FCV variants. This approach is easy to interpret and exhibits high sensitivity and specificity, making it a promising alternative for rapid detection in resource-limited settings. Furthermore, by incorporating current streamlined procedures with automated nucleic acid extraction systems, the turnaround time for infield detection can be further reduced to approximate 50 min, thereby meeting the needs of friendly operation for veterinary practitioners. Similar to other molecular methodologies, there will exist a potential risk of off-target detection and subsequent false results due to non-specific amplification and sequence mismatch in clinical specimens [29]. Therefore, meticulous design and selection of oligonucleotides for dual RPA reaction and guide RNA recognition are imperative to ensure accurate diagnosis of the FHV-1and FCV in the one-tube assay.
Notably, the preliminary one-tube assay simultaneously generated detectable signals for both targets without cross-talk, thereby demonstrating the relatively independent detection machinery of Cas12a-crRNA and Cas13a-crRNA complexes in current one-tube reaction system. However, balanced fluorescence signals for both targets were not consistently achieved in unoptimized reaction systems. Previous researchers have endeavored to orchestrate constitutive determinants to enhance the efficiency of multiplex detection, whereas the reaction parameters and components varied in diverse CRISPR/Cas-powered detection systems [23,24,25, 30]. These findings prompt us to further optimize the reaction settings for current one-tube dRPA-Cas12a/Cas13a assay targeting FHV-1 TK gene and FCV ORF1 gene.
As previously reported, the trans-cleavage activity of Cas12a or Cas13a exhibited substrate-dependent activities regarding the amounts of target templates [31]. Therefore, the optimal amount of dual RPA products was determined in present study to maximize detection efficiency for both targets. Additionally, it has been reported that crucial components involved in RNA transcription for the Cas13a-based assay could hinder the capability of the Cas12a-based assay [23, 32], so the optimal concentrations of RNase inhibitor and T7 RNA polymerase mix were also screened for current one-tube reaction systems. Unexpectedly, the remarkable inhibition conferred by RNase inhibitor on Cas12a trans-cleavage activity was not consistently observed when its amount exceeded 24 U in the 30 μL of reaction system, possibly indicating a narrow spectrum of concentration-dependent effect for RNase inhibitor, which also requires further study. Based on our previous findings [16, 17], other essential components, such as NTPs, NEBuffer, crRNA, ssDNA, and ssRNA reporters, have been optimized for efficient single RPA/Cas12a-based or RPA/Cas13a-based assay, thus they were kept at fixed amounts in current one-tube dRPA-Cas12a/Cas13a assay. Consequently, high detection performance of hybrid dRPA-Cas12a/Cas13a assay was susccessfully achieved.
Accumulative evidences have revealed that single RPA/Cas12a-based or RPA/Cas13a-based assay can effectively reduce a risk of false-positive detection due to nonspecific isothermal amplification for RPA, thereby achieving high specificity [16, 17, 30, 31]. Accordingly, the current hybrid dRPA-Cas12a/Cas13a assay showed similar advantage in accurate discrimination for specific targets, and achieved high detection sensitivity for both targets in single copy, which is lower than that of multiplex real-time PCR methods [10, 12], NanoPCR method (Nanoparticles assisted PCR) [13], and dual-ERA method (Enzymatic Recombinase Amplification) targeting FHV-1 and FCV [33]. Furthermore, the detection capabilities targeting FHV-1 and FCV were not compromised in the hybrid dRPA-Cas12a/Cas13a assay compared to single RPA/Cas12a-based or RPA/Cas13a-based assay, respectively [16, 17]. In clinical scenarios, several suspected positive samples (Ct value between 36 and 38) were identified by the dRPA-Cas12a/Cas13a assay, and the results were further confirmed by DNA sequencing. However, it should be noted that setting a rigorous cut-off Ct value (below 36) may result in unexpected discordance between dRPA-Cas12a/Cas13a assay and reference real-time PCR methods. In this case, a well-established verification technique should be introduced to discriminate true or false positives when developing a novel methodology. Moreover, discordant results for several clinical samples yielded by LDF-based assay were observed in comparison with Fluor-based assay, which might be attributed to the interference by complex compounds in samples or the possible “hook effect” of colloidal gold-based colormetric modality [34], suggesting the prudent use and interpreation of LDF-based readouts for clinical detecion. In spite of this, exceptional agreement for comparative study indicates high detection performance of dRPA-Cas12a/Cas13a assay, which is reliable and adaptable for accurate diagnosis of FHV-1 and FCV infection in clinical setting.
Although several closed-tube reaction systems have been developed as contamination-free alternatives for single Cas12a-based or Cas13a-based assay [35, 36], which mechanically separate RPA reaction system and CRISPR-Cas reaction system by built-in tube or tube lid, there still exists a concern about the compromised detection capability due to possible thermal instability of constitutive components for Cas12a-based orCas13a-based reaction system that are pre-incubated at 37~39 ℃ for another 20 min in two-step procedure. In addition, a one-step procedure was also applied as another alternative approach, whereas reduced detection capability for Cas12a-based or Cas13a-based assay was observed [36, 37]. This phenomenon may be attributed to the rapid consumption of target templates provided by RPA reaction at initial stage, which will conversely reduce the activities of CRISPR/Cas nucleases [37]. Therefore, it would be wise to deploy a strategy based on rate-limiting crRNA recognition and delayed trans-cleavage activation of CRISPR/Cas nucleases [38]. Overall, these findings suggest thatthe two-step procedure is currently a reliable approach for developing one-tube dRPA-Cas12a/Cas13a assay.
However, there are still some limitations to this study. Firstly, the open-tube procedure may lead to nucleic acids contamination, particularly in small clinical lab.Secondly, further study is required to fulfill the discrimination among herpesvirus or calicivirus lineages originating from other species. Moreover, to accomplish cost-effective and massive in-field detection, it would be beneficial to employ highly integrative approaches with low-cost workflows and high-throughput platforms, such as nucleic acid extraction-free techniques [39], amplification-free techniques [40], and microfluidic platforms [41], which can further simplify the procedures and improve the efficiency of duplex detection.
Conclusion
Altogether, the one-tube dRPA-Cas12a/Cas13a assay enables simultaneously ultrasensitive and visual detection of FHV-1 and FCV nucleic acids with user-friendly modality, which provides unparalleled convenience for FHV-1 and FCV co-infection surveillance and decision-making of URTD management.
Methods
Recombinant plasmids construction
Nucleic acids of FHV-1 and FCV are prepared using the Tiangen DNA/RNA Isolation Kit (Tiangen, Beijing, China). RNAs are reversely transcribed into cDNAs with PrimeScript™ RT reagent Kit (Takara, Dalian, China) according to the manufacturer’s instructions. The recombinant plasmid DNAs containing inserted target fragments of the FHV-1 TK gene or FCV ORF1 gene are constructed and amplified using established PCR methods, pMDTM19-T Vector (TA) Cloning Kit (Takara, Dalian, China) and E. coli DH5α Competent Cells (Takara, Dalian, China) as previouly described [16,17,18].
Primers and oligonucleotides preparation
Conservative target fragments of the FHV-1 TK gene and FCV ORF1 gene are screened by genome sequences alignment (GenBank: no. MT813047, MW194990) to design PCR, RPA primers (adding T7 promoter for RNA transcription) and CRISPR RNA (crRNA) oligonucleotides accordingly using Primer Premier 5.0 (PREMIER Biosoft, San Francisco, USA). The crRNA oligonucleotides containing T7 promoter are annealed into dsDNA using an annealing buffer for DNA oligos (Beyotime, Shanghai, China) and HiScribe T7 RNA synthesis kit (NEB, Beijing, China). Then, the synthetic crRNA is digested with DNase I (NEB, Beijing, China) at 37 °C for 15 min and purified with Monarch RNA Purification Kit (NEB, Beijing, China) according to the instruction manual. The concentration of synthetic crRNA is determined using a Qubit 3.0 fluorometer (ThermoFisher Scientific, Waltham, USA). The ssDNA and ssRNA reporters are designed and prepared according to the trans-cleavage preference of LbaCas12 (targeting FHV-1 TK gene) and LwaCas13a (targeting FCV ORF1 gene), respectively. All the primers and oligonucleotide sequences, and recognition sites are shown in Fig. 5 and supplementary Tables 1, and synthesized by Sangon Biotech (Shanghai, China).

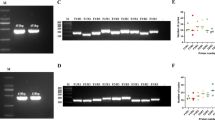
Target sequences recognized by primers and oligonucleotides. The recognition sites of RPA primers, crRNA oligonucleotides, and the reference real-time PCR primers for highly conservative FHV-1 TK gene (66,744 ∼ 66,898 nt, CH-B strain genome) and FCV ORF1 gene (2418 ∼ 2550 nt, SMU-B5-2020 strain genome) are illustrated, respectively. The TK and OFR-1 gene fragments are cloned into pMDTM19-T plasmid for later target DNAs preparation
Dual RPA reaction
To prepare target nucleic acids, the 50 μL of dual RPA (dRPA) reaction system consists of 1 μL of FHV-1 TK gene template, 1μL of FCV ORF1 gene template, 1.6μL of each RPA-TK-F/R primer (10 μM), 1.6μL of each RPA-ORF1-F/R primer (10 μM), 29.5μL of Primer Free Rehydration buffer (TwistDx, Maidenhead, UK), and 12.8 μL of DNase/RNase-Free Distilled Water (ThermoFisher Scientific, Beijing, China), then 2.5 μL of magnesium acetate (280 mM) is added according to manufacturer’s instructions of TwistAmp® Basic Kit (TwistDx, Maidenhead, UK), and the reaction tube is incubated for 20 min at 39 °C. The RPA products are purified using the UNIQ-10 Spin Column Oligo DNA Purification Kit (Sangon Biotech, Shanghai, China) and determined by gel electrophoresis.
Hybrid dRPA-Cas12a/Cas13a assay
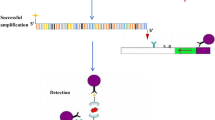
The hybrid Cas12a/Cas13a is constructed using two-step strategy, described as one-tube dRPA-Cas12a/Cas13a assay, which combine dual RPA products with hybrid CRISPR-Cas12a/Cas13a trans-cleavage activities in a single tube reaction system (Fig. 6 and Supplementary Fig. 8). In the preliminary integrated assay, the reaction system (30 μL in total) consists of 2 μL of dRPA amplicons (initial templates was 104 copies/μL), 30 nM LbaCas12a (NEB, Beijing, China), 3 μL of TK-crRNA (30 nM final), 50 nM LwaCas13a (generated as previously described) [16], 3 μL of ORF1-crRNA(45 nM final), 1.2 μL of RNase inhibitor (40 U/μL) (NEB, Beiing, China), 3 μL of T7 RNA Polymerase mix (50 U/μL) (NEB, Beijing, China), 3 μL of RNAPol Reaction Buffer (10×) (NEB, Beijing, China), 1.25 mM NTP Buffer Mix (NEB, China), 240 nM ssDNA-Cy5/BHQ3 reporter (TTATT) and 240 nM ssRNA-FAM/BHQ1 reporter (poly-14U) and complementary reaction buffer (60 mM NaCl, 40 mM Tris-HCl, 6 mM MgCl2, pH 7.3). Then, the integrative reaction system is incubated for 30 min at 37 °C, and endpoint raw fluorescence values are recorded after 30 min in CFX-96 Real-time PCR System (Bio-Rad, Hercules, USA). Subsequently, the reaction tubes are put under the Bio-Rad ChemiDoc MP imaging system (Bio-Rad, Hercules, USA) with its built-in 485 nm (λex) and 643 nm (λex) channels respectively for fluorescence readouts. For the lateral flow dipstick (LFD) readouts, HybriDetect Dipstick 2T (Milenia Biotec, Gießen, Germany) is dipped into the reaction buffer with 20 μL reaction system products containing 200 nM ssDNA-FAM/Biotin reporter (TTATT), 200 nM ssRNA-FAM/Digoxigenine reporter (poly-14U) plus 100 μL hybrid detect assay buffer, and incubated 5 min at room temperature. It is noted that the invisible colorimetric band is determined as positive for the target, and the visible band is determined as negative for the target. The further explanation of LFD readouts is shown in Supplementary Fig. 8.

Workflow of two-step one-tube dRPA-Cas12a/Cas13a assay. (A) The nucleic acid extracts are prepared from swab samples collected from feline patients with URTD. Reverse transcription is implemented for RNA templates preparation in an independent tube. After that, the target templates are transferred to another tube for immediate duplex RPA reaction (the first step). Then, the prepared dRPA products are added into the premixed hybrid Cas12-crRNA and Cas13a-crRNA complex to trigger the one-tube reaction system (the second step). Before that, the target FCV-ORF1 cDNAs are synchronously transcribed to RNAs for subsequent crRNA recognition using T7 RNA polymerase. In the one-tube hybrid reaction system, Cas12a/crRNA/target DNA oligonucleotides triplets or Cas13a/crRNA/target RNA oligonucleotides triplets are assembled, respectively. Then, hybrid LbaCas12 and (or) LwaCas13a trans-cleavage activities are triggered immediately once specific crRNA-guided recognition of target FHV-1 and (or) FCV templates in the reaction system. Subsequently, the ssDNA and ssRNA reporters are cleaved to generate visible signals. (B) Four types of visual readouts based on the dRPA-Cas12a/Cas13a assay are displayed by multi-channel fluorescence detectors or lateral flow dipsticks
Considering the possible interference between Cas12a and Cas13a activities in the one-tube system, the crucial components including amounts of dRPA products, RNase inhibitor, and T7 RNA polymerase mix were screened as independent variables, respectively. The visual readouts under the fluorescence detector were quantitatively evaluated to determine the optimal conditions for balanced and maximal fluorescence signals in both channels.
Specificity and sensitivity testing
For specificity testing, common microorganisms that may cause feline URTD are detected using the one-tube dRPA-Cas12a/Cas13a assay. FCV (SMU-B5-2020, Genbank: no. MW194990), FHV-1 (SMU-2021, sequence not submitted to GenBank), FPV (F-C, Genbank: no. OL547731.1), feline-origin Escherichia coli (E. coli) (SMU-S120, sequence not submitted to GenBank) and Klebsiella pneumoniae (K. pneumoniae) (SMU-S-10, sequence not submitted to GenBank) isolates are provided by Key Laboratory of Animal Medicine at Southwest Minzu University of Sichuan Province. Positive samples of Mycoplasma felis (M. felis), Chlamydia felis (C. felis), Bordetella bronchiseptica (B. bronchiseptica), and FCoV are confirmed by a commercial real-time PCR kit (Glinx, Shanghai, China). Then, the DNAs or RNAs of these samples are tested using the one-tube assay.
For sensitivity testing, serial dilutions of plasmid DNAs (107∼10–2 copies/μL) with equal amounts of FHV-1 and FCV templates are detected by current dRPA-Cas12a/Cas13a assay and the reference real-time PCR methods [17, 18]. The primers and oligonucleotides sequences used in this study and their matching sites along the virus genome are illustrated in Fig. 5. The dsDNA copy number is determined using the following formula: {[6.02 × 1023×dsDNA concentration(ng/μL)×10− 9]}/[DNA in length×660].
Validation of one-tube dRPA-Cas12a/Cas13a assay in clinical samples
Fifty-six conjunctive, nasal and oropharyngeal swabs were collected from cats with URTD, who visited the Veterinary Hospital of Southwest Minzu University, Chengdu City, China, in 2021. The samples are maintained in RNALater™ Viral RNA stable preservation solution (Beyotime, Shanghai, China) for later processing. Then, the nucleic acids of samples are prepared and tested using the dRPA-Cas12a/Cas13a assay (Fluor-based assay and LDF-based assay) and the reference real-time PCR methods, respectively. The real-time PCR is performed on the CFX-96 Real-time PCR System (Bio-Rad, Hercules, USA) using TB Green Premix Ex Taq (Takara, Dalian, China) [16, 18]. The cycle threshold (Ct) values below 36 are determined as the cut-off value for positive results. The Ct values between 36 and 38 are for suspected positive results. The discordant samples between dRPA-Cas12a/Cas13a assays and reference real-time PCR tests are further identified by Sanger sequencing (Sangon Biotech, Shanghai, China).
Statistical data analysis
Fluorescence readouts and lateral flow dipstick readouts (Supplementary Fig. 8) are photographed by camera and automatically adjusted using ImageJ (NIH, USA). The statistical differences of fluorescence variables are analyzed using the Wilcoxon test or Mann Whitney U test in Prism 8.0 (GraphPad Software Inc, USA). The consistency analysis (sensitivity, specificity, and Kappa value) between the dRPA-Cas12a/Cas13a assay and reference real-time PCR methods is achieved using SPSS 18.0 (IBM, USA).
Data availability
The data and materials supporting the findings of current study are available in the article and supplementary files.
Abbreviations
- B. bronchiseptic :
-
Bordetella bronchiseptica
- C. felis :
-
Chlamydia felis
- Cas:
-
CRISPR associated proteins
- CRISPR:
-
Clustered regularly interspaced palindromic repeats
- crRNA:
-
CRISPR RNA
- E. coli :
-
Escherichia coli
- FCoV:
-
Feline coronavirus
- FCV:
-
Feline calicivirus
- FHV-1:
-
Feline herpesvirus type 1
- FPV:
-
Feline panleukopenia virus
- K. peumoniae :
-
Klebsiella pneumoniae
- M. felis :
-
Mycoplasma felis
- RPA:
-
Recombinase polymerase amplification
- RT-qPCR:
-
Real time quantitative polymerase chain reaction
- URTD:
-
Upper respiratory tract diseases
References
Helps CR, Lait P, Damhuis A, Björnehammar U, Bolta D, Brovida C, Chabanne L, Egberink H, Ferrand G, Fontbonne A, Pennisi MG, Gruffydd-Jones T, Gunn-Moore D, Hartmann K, Lutz H, Malandain E, Möstl K, Stengel C, Harbour DA, Graat EAM. Factors associated with upper respiratory tract disease caused by feline herpesvirus, feline calicivirus, Chlamydophila felis and Bordetella bronchiseptica in cats: experience from 218 European catteries. Vet Rec. 2005;156(21):669–73. https://doi.org/10.1136/vr.156.21.669
Palombieri A, Di Profio F, Fruci P, Sarchese V, Martella V, Marsilio F, Di Martino B. Emerg Respiratory Viruses Cats Viruses. 2022;14(4):663. https://doi.org/10.3390/v14040663
André NM, Miller AD, Whittaker GR. Feline infectious peritonitis virus-associated rhinitis in a cat. JFMS Open Rep. 2020;6(1):2055116920930582. https://doi.org/10.1177/2055116920930582
Mavrides DE, Morgan AL, Na JG, Graham PA, McHugh TD. Antimicrobial resistance profiles of bacteria associated with lower respiratory tract infections in cats and dogs in England. Vet Rec. 2022;190(4):e779. https://doi.org/10.1002/vetr.779
Aslam MW, Lau SF, Radzi R, Omar S, Kaka U, Ahmed I. Clinicopathological and radiological features of cats presented with infectious respiratory disease signs: a focus on Rhodococcus equi and Klebsiella pneumoniae. Microorganisms. 2023;11(3):737. https://doi.org/10.3390/microorganisms11030737
Litster A, Wu CC, Leutenegger CM. Detection of feline upper respiratory tract disease pathogens using a commercially available real-time PCR test. Vet J. 2015;206:149–53. https://doi.org/10.1016/j.tvjl.2015.08.001
Fernandez M, Manzanilla EG, Lloret A, León M, Thibault JC. Prevalence of feline herpesvirus-1, feline calicivirus, Chlamydophila felis and Mycoplasma felis DNA and associated risk factors in cats in Spain with upper respiratory tract disease, conjunctivitis and/or gingivostomatitis. J Feline Med Surg. 2017;19:461–9. https://doi.org/10.1177/1098612X16634387
Townsend WM, Jacobi S, Tai SH, Kiupel M, Wise AG, Maes RK. Ocular and neural distribution of feline herpesvirus-1 during active and latent experimental infection in cats. BMC Vet Res. 2013;22:9185. https://doi.org/10.1186/1746-6148-9-185
Berger A, Willi B, Meli ML, Boretti FS, Hartnack S, Dreyfus A, Lutz H, Hofmann-Lehmann R. Feline calicivirus and other respiratory pathogens in cats with feline calicivirus-related symptoms and in clinically healthy cats in Switzerland. BMC Vet Res. 2015;11:282. https://doi.org/10.1186/s12917-015-0595-2
Hussein ITM, Field HJ. Development of a quantitative real-time TaqMan PCR assay for testing the susceptibility of feline herpesvirus-1 to antiviral compounds. J Virol Methods. 2008;152(1–2):85–90. https://doi.org/10.1016/j.jviromet.2008.05.018
Abd-Eldaim MM, Wilkes RP, Thomas KV, Kennedy MA. Development and validation of a TaqMan real-time reverse transcription-PCR for rapid detection of feline calicivirus. Arch Virol. 2009;154(4):555–60. https://doi.org/10.1007/s00705-009-0337-5
Cao N, Tang ZH, Zhang XY, Li WY, Li BX, Tian YB, Xu DN. Development and application of a triplex TaqMan quantitative real-time PCR assay for simultaneous detection of Feline Calicivirus, Feline Parvovirus, and Feline Herpesvirus. Front Vet Sci. 2021;8:792322. https://doi.org/10.3389/fvets.2021.792322
Ye JF, Li ZJ, Sun FY, Guo Li, Feng EK, Bai X, Cheng YN. Development of a triple NanoPCR method for feline calicivirus, feline panleukopenia syndrome virus, and feline herpesvirus type I virus. BMC Vet Res. 2022;18(1):379. https://doi.org/10.1186/s12917-022-03460-9
Wang JC, Liu LB, Wang JF, Sun XX, Yuan WZ. Recombinase polymerase amplification Assay-A simple, fast and cost-effective alternative to real time PCR for specific detection of Feline Herpesvirus-1. PLoS ONE. 2017;12:e0166903. https://doi.org/10.1371/journal.pone.0166903
Liu D, Zheng YT, Yang YP, Xu XY, Kang HT, Jiang Q, Yang MF, Qu LD, Liu JS. Establishment and application of ERA-LFD method for rapid detection of feline calicivirus. Appl Microbiol Biotechnol. 2022;106:1651–61. https://doi.org/10.1007/s00253-022-11785-6
Huang J, Liu YJ, He YW, Yang XN, Li Y. CRISPR-Cas13a based visual detection assay for Feline Calicivirus circulating in Southwest China. Front Vet Sci. 2022;9:913780. https://doi.org/10.3389/fvets.2022.913780
Huang J, Liu YJ, Yang XN, Li Y. RPA-Cas12a-LFD based nucleic acid detection for Feline Herpesvirus-1 and preliminary application (in Chinese). Acta Vet Et Zootechnica Sinica. 2022;53:1638–43. https://doi.org/10.11843/j.issn.0366-6964.2022.05.032
Helps CR, Lait P, Tasker S, Harbour D. Melting curve analysis of feline calicivirus isolates detected by real-time revereaction systeme transcription PCR. J Virol Methods. 2002;106:241–4. https://doi.org/10.1016/s0166-0934(02)00167-2
Chen JS, Ma E, Harrington LB, Costa MD, Tian XR, Palefsky JM, Doudna JA. CRISPR-Cas12a target binding unleashes indiscriminate single-stranded DNase activity. Science. 2018;360:436–9. https://doi.org/10.1126/science.aar6245
Gootenberg JS, Abudayyeh OO, Lee JW, Essletzbichler P, Dy AJ, Joung J, Verdine V, Donghia N, Daringer NM, Freije CA, Myhrvold C, Bhattacharyya RP, Livny J, Regev A, Koonin EV, Hung DT, Sabeti PC, Collins JJ, Zhang F. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science. 2017;356:438–42. https://doi.org/10.1126/science.aam9321
Gootenberg JS, Abudayyeh OO, Kellner MJ, Joung JL, Collins JJ, Zhang F. Multiplexed and portable nucleic acid detection platform with Cas13, Cas12a, and Csm6. Science. 2018;360(6387):439–44. https://doi.org/10.1126/science.aaq0179
Xiong EH, Jiang L, Tian T, Hu ML, Yue HH, Huang MQ, Lin W, Jiang YZ, Zhu DB, Zhou XM. Simultaneous dual-gene diagnosis of SARS-CoV-2 based on CRISPR/Cas9-mediated lateral flow assay. Angew Chem. 2021;60:5307–15. https://doi.org/10.1002/anie.202014506
Tian T, Qiu ZQ, Jiang YZ, Zhu DB, Zhou XM. Exploiting the orthogonal CRISPR-Cas12a/Cas13a trans-cleavage for dual-gene virus detection using a handheld device. Biosens Bioelectron. 2022;196:113701. https://doi.org/10.1016/j.bios.2021.113701
Su GX, Zhu M, Li DY, Xu MT, Zhu YD, Zhang Y, Zhu HY, Li F, Yu YY. Multiplexed lateral flow assay integrated with orthogonal CRISPR-Cas system for SARS-CoV-2 detection. Sens Actuators B Chem. 2022;371:132537. https://doi.org/10.1016/j.snb.2022.132537
Li LN, Duan CX, Weng JF, Qi XT, Liu CL, Li XH, Zhu JJ, Xie CX. A field-deployable method for single and multiplex detection of DNA or RNA from pathogens using Cas12 and Cas13. Sci China Life Sci. 2022;65:1456–65. https://doi.org/10.1007/s11427-021-2028-x
Cao GH, Dong JB, Chen XL, Lu P, Xiong YF, Peng L, Li JW, Huo DQ, Hou CJ. Simultaneous detection of CaMV35S and T-nos utilizing CRISPR/Cas12a and Cas13a with multiplex-PCR (MPT-Cas12a/13a). Chem Commun. 2022;58:6328–31. https://doi.org/10.1039/d2cc01300b
Ding LH, Wu Y, Liu LE, He LL, Yu SC, Effah CY, Liu X, Qu LB, Wu YJ. Universal DNAzyme walkers-triggered CRISPR-Cas12a/Cas13a bioassay for the synchronous detection of two exosomal proteins and its application in intelligent diagnosis of cancer. Biosens Bioelectron. 2022;219:114827. https://doi.org/10.1016/j.bios.2022.114827
Zheng X, Li YK, Yuan MZ, Shen Y, Chen SY, Duan GC. Rapid detection of HPV16/18 based on a CRISPR-Cas13a/Cas12a dual-channel system. Anal Methods. 2022;14(48):4607–16. https://doi.org/10.1007/s00253-022-12015-9
Daher RK, Stewart G, Boissinot M, Boudreau DK, Bergeron MG. Influence of sequence mismatches on the specificity of recombinase polymerase amplification technology. Mol Cell Probes. 2015;29:116–21. https://doi.org/10.1016/j.mcp
Zhu YT, Xing C, Yang L, Li Q, Wang XF, Zhou J, Zhang C, Ren CP, Liu FH, He J, Shen B, Du YN, Liu Y. Dual-gene detection in a single-tube system based on CRISPR-Cas12a/Cas13a for severe fever thrombocytopenia syndrome virus. Front Microbiol. 2022;13:977382. https://doi.org/10.3389/fmicb.2022.977382
Nalefski EA, Patel N, Leung PJ, Islam Z, Kooistra RM, Parikh I, Marion E, Knott GJ, Doudn A, Anne-Laure JA, Ny ML, Madan D. Kinetic analysis of Cas12a and Cas13a RNA-guided nucleases for development of improved CRISPR-based diagnostics. iScience. 2021;24:102996. https://doi.org/10.1016/j.isci.2021.102996
Ding X, Yin K, Li ZY, Rajesh VL, Enrique B, Maroun MS, Liu CC. Ultrasensitive and visual detection of SARS-CoV-2 using all-in-one dual CRISPR-Cas12a assay. Nat Commun. 2020;11(1):4711. https://doi.org/10.1038/s41467-020-18575-6
Chen B, Zhang HY, Wang HH, Li SJ, Zhou P. Development and application of a dual ERA method for the detection of Feline Calicivirus and Feline Herpesvirus Type I. Virol J. 2023;20:62. https://doi.org/10.1186/s12985-023-02020-3
Ross GMS, Filippini D, Nielen MWF, Salentijn GI. Unraveling the hook effect: a comprehensive study of high antigen concentration effects in sandwich lateral flow immunoassays. Anal Chem. 2020;92(23):15587–95. https://doi.org/10.1021/acs.analchem.0c03740
Hu F, Liu YF, Zhao SH, Zhang ZM, Li XC, Peng NC, Jiang ZD. A one-pot CRISPR/Cas13a-based contamination-free biosensor for low-cost and rapid nucleic acid diagnostics. Biosens Bioelectron. 2022;202:113994. https://doi.org/10.1016/j.bios.2022.113994
Xiong YF, Cao GH, Chen XL, Yang J, Shi MM, Wang Y, Nie FP, Huo DQ, Hou CJ. One-pot platform for rapid detecting virus utilizing recombinase polymerase amplification and CRISPR/Cas12a. Appl Microbiol Biotechnol. 2022;106(12):4607–16. https://doi.org/10.1007/s00253-022-12015-9
An BL, Zhang HB, Su X, Guo Y, Wu T, Ge YY, Zhu FF, Cui LB. Rapid and sensitive detection of Salmonella spp. Using CRISPR-Cas13a Combined With Recombinase Polymerase Amplification. Front Microbiol. 2021;12:732426. https://doi.org/10.3389/fmicb.2021.732426. eCollection 2021.
Hu ML, Bi ZQ, Tian ZR, Jiang T, Zhou YZ. Photocontrolled crRNA activation enables robust CRISPR-Cas12a diagnostics. Proc Natl Acad Sci. 2022;119:e2202034119. https://doi.org/10.1073/pnas.2202034119
Azmi I, Faizan MI, Kumar R, Yadav SR, Chaudhary N, Singh DK, Butola R, Ganotra A, Joshi GD, Jhingan GD, Iqbal J, Joshi MC, Ahmad T. eCollection. A saliva-based rna extraction-free workflow integrated with Cas13a for SARS-CoV-2 detection. Front Cell Infect Microbiol.2021,11:632646. https://doi.org/10.3389/fcimb.2021.632646. 2021.
Shinoda H, Taguchi Y, Nakagaw R, Makino A, Okazaki S, Nakano M, Muramoto Y, Takahashi C, Takahashi I, Ando J, Noda T, Nureki O, Nishimasu H, Watanabe R. Amplification-free RNA detection with CRISPR-Cas13. Commun Biol. 2021;4(1):476. https://doi.org/10.1038/s42003-021-02001-8
Welch NL, Zhu ML, Hua C, Weller J, Mirhashemi ME, Nguyen TG, Mantena S, Bauer MR, Shaw BM, Ackerman CM, Thakku SG, Tse MW, Kehe J, Uwera MM, Eversley JS, Bielwaski DA, McGrath G, Braidt J, Johnson J, Cerrato F, Moreno GK, Krasilnikova LA, Petros BA, Gionet GL, Huard EK, Jalbert RC, Cleary SK, Fitzgerald ML, Gabriel NA, Gallagher SB, Smole GR, Madoff SC, Brown LC, Keller CM, Wilson MW, Kirby MM, Barnes MK, Park JR, Siddle DJ, Happi KJ, Hung CT, Springer DT, MacInnis M, Lemieux BL, Branda JEER, Blainey JA, Sabeti PC, Myhrvold PC. Multiplexed CRISPR-based microfluidic platform for clinical testing of respiratory viruses and identification of SARS-CoV-2 variants. Nat Med. 2022;28:1083–94. https://doi.org/10.1038/s41591-022-01734-1
Acknowledgements
We acknowledge the collaborators in Veterinary Teaching Hospital of Southwest Minzu University who provided the clinical samples, and appreciate Huijun Long and James Adams from University of Surrey, United Kingdom, for writing revision.
Funding
The study was supported by “The Fundamental Research Funds for the Central Universities, Southwest Minzu University” (Grant No. ZYN2023046).
Author information
Authors and Affiliations
Contributions
FmJ: writing-original draft, data sorting, YjL: methodology establishment, data sorting and analysis. XnY: collaborated in the writing, analysis and interpretation of data. YL: study design, analysis, manuscript writing and revision. JH: funding acquisition and writing review and editing. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Animal ethics was approved by the Research Ethics Committee of Southwest Minzu University (approval number No.2021-MDLS-028). Informed consent was obtained from cat owners visiting to Veterinary Teaching Hospital of Southwest Minzu Univeristy. Sampling and handling for samples collection followed the criteria of animal welfare regulations. All of our methods were complied with the statement of the ARRIVE guidelines report and were carried out in accordance with relevant guidelines and regulations.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jiang, F., Liu, Y., Yang, X. et al. Ultrasensitive and visual detection of Feline herpesvirus type-1 and Feline calicivirus using one-tube dRPA-Cas12a/Cas13a assay. BMC Vet Res 20, 106 (2024). https://doi.org/10.1186/s12917-024-03953-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-024-03953-9