
Abstract
Background
Multidrug resistance in Enterobacteriaceae including resistance to quinolones is rising worldwide. The development of resistance may lead to the emergence of new transmission mechanisms. In this study, the collection of different E. coli was performed from animals and subjected to subsequent procedures including pulsed-field gel electrophoresis, micro-broth dilution method, polymerase chain reaction. Whole genome sequencing of E. coli C3 was performed to detect the affinity, antimicrobial resistance and major carriers of the isolates.
Results
A total of 66 E. coli were isolated and their antibiotic resistance genes, frequency of horizontal transfer and genetic environment of E. coli C3 were determined. The results showed there were both different and same types in PFGE typing, indicating clonal transmission of E. coli among different animals. The detection of antimicrobial resistance and major antibiotic resistance genes and the plasmid transfer results showed that strains from different sources had high levels of resistance to commonly used clinical antibiotics and could be spread horizontally. Whole-genome sequencing discovered a novel ICE mobile element.
Conclusion
In summary, the antimicrobial resistance of E. coli in northeast China is a serious issue and there is a risk of antimicrobial resistance transmission. Meanwhile, a novel ICE mobile element appeared in the process of antimicrobial resistance formation.
Similar content being viewed by others


Background
Antimicrobial resistance significantly threatens public health [1]. Multidrug resistance (MDR) pathogens are responsible for at least 700,000 deaths yearly [2]. E. coli is an important vector and potential source for the transmission of antibiotic resistance genes (ARGs) [3, 4], which are commonly transmitted to other bacteria in humans during breeding, slaughtering, and processing [5, 6]. E. coli has an important role in the spread of antimicrobial resistance (AMR) as a donor, recipient, and intermediate carrier of ARGs [7, 8]. In addition, bacteria can capture foreign genes through mobile elements such as mobile plasmids, integrons, mobile transposons, Integrative and Conjugative Elements (ICE), etc. [9, 10], and integrate and regulate ARGs, thereby obtaining the horizontal transfer of AMR. Therefore, the resistance carried by E. coli is more prone to horizontal transmission [9]. Previous studies have also reported that overuse of antibiotics, great number of animals, and low genetic diversity from intensive farming increase the risk of animal pathogens transferring to humans. For example, many kinds of E. coli have been detected in veterinary clinics, but their epidemic characteristics are mainly found in some intensive farms. E. coli not only caused certain economic losses to animal husbandry but also seriously threatened the public health safety of human beings [11].
In recent years, the antibiotic resistance of some strains, such as methicillin-resistant Staphylococcus [12], carbapenem-resistant Enterobacteriaceae [13], and MDR Acinetobacter baumannii [14], have attracted increasing attention. Recently, the emergence of tet(X4) [15] and mcr-1 [16] have come to pose a threat to the clinical utility of tigecycline and colistin, the last line of defense for humans.
ICE is a novel mobile element found in bacteria that can transfer between bacteria through conjugation. Many ICEs are associated with ARGs; transferring these genetic elements can accelerate the dissemination of ARGs within and between microbial genera [17]. ICE has some relatively conserved core genes, including int, xis and tra, whose ends are generally inverted repeats attL and attR. Also, ICE has a typical modular structure that can be divided into four modules according to the different functions mediated by gene clusters: integration and excision module (int-xis), mating-pair formation module (mpf), mobilization and processing (mob), and regulation (reg). In addition, ICE also contains some hot spots (HS) and variable regions (VR) foreign genes [18]. In 2002, complex elements that can be cut from the chromosome, integrated into the host chromosome, and transferred between bacterial cells by conjugation were first classified as ICE [19]. In the study of gram-negative intestinal bacteria, ICE R391 has raised concerns. It was found in Providencia reitseri, isolated from human excreta in South Africa in 1967 and initially called IncJ plasmid [20]. However, genomic analysis suggests that R391 is an ICE some 89 kb long. A large type of ICE that is more concerned by various studies is SXT [21].
The ARGs of ICE have some association with transposons, and some ICE contain complex transposon structures composed of IS16 or IS26, which surround the ARGs [22]. In their study, Beaber et al. found that the widespread use of quinolone antibiotics increases the frequency of associated ICE transfer [23]. As a result, the frequency of ICE-carrying bacteria in the clinic and the environment gradually increases. Consequently, Bi and his team created ICEberg (http://db-mml.sjtu.edu.cn/ICEberg/), an integrated conjugation element database, which includes 428 ICEs in 333 bacterial strains identified through bioinformatics prediction and literature mining [24].
The World Health Organization (WHO) has classified fluoroquinolones as “critically important antimicrobials” because of their broad-spectrum effects and clinical importance in human and animal medicine [25]. Some studies have shown that minimal inhibitory concentration (MIC) ≥ 32 mg/L is a high-level resistant strain [26, 27]. In this study, an MDR E. coli C3 highly resistant to multiple antibiotics was isolated from E. coli isolated from different animals in northeast China. Whole genome analysis showed that this strain carries multiple ARGs and the novel ICE mobile element (using the database created by Bi et al.), which could be actively transmitted between bacteria through plasmids and ICE.
Results
Separation and identification
The Molecular Evolutionary Genetics Analysis (MEGA6.0) software was used to analyze the evolution based on the 16s RNA sequences of E. coli and standard strain E. coli ATCC® 25,922™ isolated from different animal sources. The results are shown in Fig. 1a. Through the construction and analysis of the evolutionary tree, 66 E. coli were classified into two large branches, among which different animals as a source for E. coli belonged to the same branch and showed high homology.

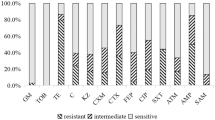
Phylogenetic tree of E. coli isolates and AMR detection. (a) Phylogenetic tree of 66 E. coli. (b) The result of antimicrobial susceptibility testing. FFC, florfenicol; PB, polymyxin B; MEM, meropenem; TGC, tigecycline; MAR, marbofloxacin; TE, tetracycline; AZM, azithromycin; ENR, enrofloxacin; CN, gentamicin; CRO, ceftriaxone
Resistance to antimicrobial agents
Among 66 isolates, 40 were resistant to florfenicol (FFC), 2 were resistant to polymixin B (PB), 35 were resistant to tetracycline (TE), 30 were resistant to marbofloxacin (MAR), 11 were resistant to azaerythromycin (AZN), 17 were resistant to gentamicin (CN), 30 were resistant to ceftriaxone (CRO), and 30 were resistant to enrofloxacin (ENR) (Fig. 1b). A number of MDR strains and AMR rates are shown in Fig. 2. The highest resistance rates to FFC, TE, AZM, and CRO were found in chicken (80%, 87%, 33%, and 87%, respectively); the highest resistance rates to ENR and CN were found in pigs (71% and 53%, respectively); no strains were resistant TGC and MEM; PB-resistant strains were only found in swine sources.

Statistical analysis of antimicrobial susceptibility testing results of the isolates. (a) Resistance rate of the isolates to different antimicrobial agents. (b) Distribution of MDR strains (%). FFC, florfenicol; PB, polymyxin B; MEM, meropenem; TGC, tigecycline; MAR, marbofloxacin; TE, tetracycline; AZM, azithromycin; ENR, enrofloxacin; CN, gentamicin; CRO, ceftriaxone
Pulsed-field gel electrophoresis (PFGE) typing of E. Coli
After PFGE typing of 66 E. coli, 52 strains were successfully obtained; the Xba1 enzyme could not digest 14 strains that were not typed. A total of 36%-100 bands were similar in 52 E. coli (Fig. 3). The DNA similarity of 14 clusters reached more than 85%, totaling 30 strains. In summary, 52 E. coli strains were divided into 36 types.

Cluster analysis of PFGE electrophoresis of E. coli strains. 52 E. coli strains were divided into 36 types. The DNA similarity of 14 clusters reached more than 85%, a total of 30 strains. The DNA map similarity of the remaining 22 strains was less than 85%, and they were divided into 22 types
Detection of major ARGs
The results of the major ARGs detection of 66 E. coli are shown in Fig. 4. The detection rates from high to low were as follows: tetracycline resistance genes were tet(W) (98.5%), tet(A) (84.8%), tet(B) (12.1%), and tet(M) (6.0%); aminoglycoside resistance genes were aphA1 (100%) and aadD (36.4%), aac(2’)-IC (7.6%); macrolide resistance gene was vagB (15.1%); fluoroquinolone resistance genes were qnrA (50.0%), qnrS (16.7%), and qnrD (3.1%); chloramphenicol resistance genes were floR (78.8%) and fexA (9.1%); β-lactam resistance genes were blaTEM (90.9%).

ARGs tested results of 66 isolated strains. Tetracycline resistance genes were tet(W) (98.5%), tet(A) (84.8%), tet(B) (12.1%) and tet(M) (6.0%) while tet(S) and tet(O) were not detected, aminoglycoside resistance genes were aphA1 (100%) and aadD (36.4%), aac(2’)-IC (7.6%), macrolide resistance gene was vagB (15.1%), fluoroquinolone resistance genes were qnrA (50.0%), qnrS (16.7%) and qnrD (3.1%), chloramphenicol resistance genes were floR (78.8%) and fexA (9.1%) %) while cfr and fexB were not detected, β-lactam resistance genes were blaTEM (90.9%)
Table 1 shows the target site detection results of 7 high-level fluoroquinolone-resistant strains with different PFGE type. There were two types of amino acid mutations in the test strains, namely gyrA (Ser83Leu/Asp87Asn) and parC (Ser80Ile/Glu84Ala) mutations.
Detection of class I integron and variable regions in fluoroquinolone-resistant E. Coli
Seven E. coli carried class I integrase genes, and the positive rate was 100%. The positive strains carrying class I integrons were amplified by variable regions; the amplified products were 725-1925 bp in size. Three ARGs boxes were found, including the resistance genes encoding dihydrofolate reductase (dfrA7, dfrA17), aminoglycoside (addA22 and addA1), chloramphenicol (cmlA1) and rifampicin resistance (arr-2). One of them carried the addA1, dfrA7, arr-2 and cmlA1 resistance genes at the same time, as shown in Table 2.
Plasmid transferability
After transfer by conjugation, the antimicrobial susceptibility test determined the MIC of Salmonella H9812-pN, Salmonella H9812-pY1, and Salmonella H9812-pC3, as shown in Table 3.
The conjugation transfer frequency of Y1 strain plasmid, E. coli C3 plasmid and N strain plasmid was 3.16 × 10− 3, 6.42 × 10− 2 and 4.12 × 10− 4, respectively. E. coli C3 plasmid had the highest plasmid transfer frequency by conjugation, indicating that the plasmid contained in the E. coli C3 was most prone to transfer.
The plasmids extracted by C3, N and Y1 were transferred into Stellar competent cells by electroporation. The optimal electroshock conditions optimized during the experiment were plasmid-1 µL, voltage-2400 V, resistance-200Ω, and capacitance-25 µF. The results of C3 are shown in Table 3.
Whole genome sequencing
Whole genome sequencing (WGS) information of the E. coli C3 is shown in Table S3. The strain had obvious advantages in carbohydrate transport and metabolism, amino acid transport and metabolism, DNA replication, recombination and repair, and energy production and conversion (Figure S1).
Next, we used CARD-RGI (Comprehensive Antibiotic Resistance Database -Resistance Gene Identifier) to align the amino acid sequence of E. coli C3. The results showed 71 genes related to AMR on the C3 genome, including 42 genes involved in efflux, 14 genes with altered target sites, 9 genes that produced inactivated enzymes, 2 genes involved in biofilm formation, etc. The statistical results are shown in Table S4.
Through the whole genome sequence analysis of C3, 946 virulence genes were predicted, including enterobactin entE, flagellar operon flgL, flgK and many others, and the homology was generally up to 100%.
MGE and ICE finding online database (https://db-mml.sjtu.edu.cn/ICEfinder/) in the Center for Genomic Epidemiology database was then used to compare the whole genome sequences of E. coli C3. The results showed 4 ICE in E. coli C3, contained a novel ICE (Fig. 5). The ARGs such as floR, cmlA, blaCTX−M, blaOXA10, qnrS, tet(A) and dfrA on plasmid pC3-2 formed an MDR gene cluster of about 27 kb. The related ARGs were located and found that tet(A) (GE004795), qnrS (GE004788), floR (GE004769), cmlA (GE004777), blaOXA10 (GE004778), blaCTX−M (GE004782), dfrA (GE004780), aadA1 (GE004779) and arr (GE004776) were located on this mobile element. This element also contained the insert IS26 (covering all resistance genes). Through comparison and analysis, it was found that the basic skeleton was mainly derived from the chromosome of E. coli AP022218.1.

Gene distribution and ICE sequence analysis of pC3-2 plasmid in E. coli C3. (a) A 124 kb pC3-2 plasmid contained 19 ARGs, 3 gene islands, and a novel ICE mobile element. floR, cmlA, blaCTX−M, blaOXA10, qnrS, tet(A), dfrA and other ARGs in pC3-2 constituted an MDR gene cluster of about 27 kb. (b) It was found that the basic skeleton of ICE was mainly derived from the plasmid of E. coli AP022218.1. tet(A), qnrS, floR, cmlA, blaOXA10, blaCTX−M, dfrA, aadA1 and arr were located on the mobile element; The element also contained the insertion sequence IS26 (covering all ARGs).
Discussion
E. coli of animal origin is considered an important pathogen and a causative agent for various bacterial diseases. Antimicrobial resistance (AMR) in E. coli is considered one of the major challenges in both animals and humans and is considered a real public health concern. Recent studies found that AMR is often acquired and disseminated through the movement of mobile genetic elements, including insertion sequences, transposons, integrons, and conjugative plasmids [28, 29]. In this study, E. coli was isolated from different animal sources and genotyped. The AMR analyses were performed, and resistant and virulence strains were evaluated to reflect the regional AMR status and distribution. It was found that a strain with severe resistance and a new variant gene of the ICE mobile element could mediate the horizontal spread of MDR genes.
We observed that only 21% of the isolates exhibited susceptibility to antibiotics, while 79% demonstrated resistance to at least one antibiotic, with 70% displaying MDR. Mahipal et al. found that 63.2% of the E. coli isolated in northern India were resistant to one or more antibiotics, of which 41% showed MDR [30]. However, Enne et al. found that only 5.7% of strains isolated from cattle (British slaughterhouse) were resistant to one or more antibiotics, while 92.1% of strains isolated from pig were resistant to at least one antibiotic [31]. Although we did not assess bovine samples, isolates from pigs were significantly larger than those from other animals, and no resistance was detected in the isolates from sheep. In addition, we did not detect TGC and carbapenem-resistant strains but found PB-resistant strains. Compared with Makarov’s study, 33% of colistin-resistant strains were higher than our results. Colistin is considered the last line of defense for human beings [32]. Moreover, our study showed that the resistance rate of FFC was the highest, followed by tetracyclines and fluoroquinolones, which was higher than that reported by Zhao (50.3% resistance to tetracyclines) and lower than that that reported by Makarov (88% restence to fluoroquinolones). Further focus should be placed on the resistance rate of tetracycline or fluoroquinolones [33].
The ARGs in the integron can be arranged in different combinations [34, 35]. It has been reported that the dfr (dfrA16, dfrA17) + aad (aadA1, aadA2, aadA5) gene family in the ARG boxes is the most common combination, encoding dihydrofolate reductase and aminoglyco adenosyltransferase, respectively [36]. Among them, type I integrase is the most common in MDR strains, contributing to the horizontal spread of ARGs in different genera, animals, humans and the environment [37]. Numerous studies have suggested that ARGs can be transferred through different transferable elements, such as insertion sequence (IS), transposon (Tn), integrin, plasmid, etc. [38]. Thus, in this study, conjugative transfer and electroporation experiments were performed to determine whether these resistance genes are located on the mobile plasmids. Our results showed that 3 of the 7 strains could be conjugated successfully, and E. coli C3 showed the highest plasmid conjugation transfer frequency of 6.42 × 10− 2, suggesting that the plasmids’ horizontal spread in E. coli C3 is fast and wide. Therefore, we electroporated the E. coli HST08 strain, and the MIC results were the same as those of the zygote.
An MDR E. coli C3 was highly antibiotic-resistant and was subject to horizontal gene transfer. Next, the WGS of the strain was performed to assess the cause of AMR transfer in E. coli C3. The two plasmids were typed, pC3-1 plasmid was IncFIB (similarity 98.39%) or IncFIC (similarity 95.79), and plasmid pC3-2 (Fig. 5a, Fig. S3b) was IncX1 (similarity 98.93%) [39]. Nineteen ARGs were located on the plasmid pC3-2, including floR, cmlA, blaCTX−M, blaOXA10 and qnrS, etc., which can mediate β-lactams, chloramphenicol, tetracyclines, quinolones and other antibiotics, and formed an ARG cluster of about 27 kb in the plasmid pC3-2. The novel ICE (Fig. 5c, Fig. S2, Fig. S3a) carried by the plasmid pC3-2 contained 9 ARGs, including floR, cmlA, blaCTX−M, blaOXA10, qnrS, tet(A), dfrA, aadA1, etc. The traceability analysis of the ICE showed that its main skeleton was derived from other E. coli. At the same time, the plasmid pC3-2 also carried three composite transposons IS26, ISVsa3 and IS102, and the transposon carried a variety of ARGs such as blaCTX−M, blaOXA10, floR, tet(A), aph(3)-I, etc. The transposon IS26, ICE, and ISVsa3 all had partially overlapping ARGs. IS102 carried ARGs similar to IS26 and ISVsa3. According to related studies, IncX-type plasmids, IS26, ISVsa3, IS102, and ICE, can all mediate the horizontal transfer of ARGs [40]. For example, the ubiquitous presence of the IS element, as well as the formation and transposition of a circular IS-tet(X) variant intermediate, indicates the potential translocation of the tet(X) variant genes between different plasmids and integration into the chromosome [15].
Conclusion
The E. coli from different animals in northeast China showed different levels of AMR to commonly used antibiotics, and these E. coli could be transmitted by cloning. At the gene level, these strains carry genes that could mediate MDR, among which E. coli C3 was more resistant and carried a large number of virulence genes. A novel ICE variation was found in the bacterial evolution process of E. coli C3. The existence of E. coli C3 in the animal environment poses a serious threat to the environment and even human health and safety. Under the pressure of antibiotics, bacteria could obtain plasmids of other strains to fuse with the plasmids of this strain, carrying more ARGs, and the plasmids contained multiple IS insertion elements. These findings show the importance of rational use of antibiotics.
Methods
Sample collection
A total of 280 samples were collected in northeast China from September 2019 to August 2021, including 60 samples from chicken, 60 from goose, 80 from pigs, and 80 samples from sheep. Sterile cotton swabs were used for fresh fecal sampling; each sample was sealed, labeled, and then returned to the laboratory for sample processing as soon as possible. After purification, 16s rRNA universal primers for polymerase chain reaction (PCR) amplification, PCR products link pMD18-T carrier, were used on a single colony. Samples were sent for sequencing to sangon biological engineering (Shanghai) co., LTD., sequencing results were analyzed using BLAST on https://blast.ncbi.nlm.nih.gov.
PFGE typing of E. Coli
In order to determine the genetic relatedness of the E. coli. Isolates were digested with the restriction enzyme XbaI (3µL, 15U/µL). The gels were run at 6.0 V/cm with an initial/final switch time of 2 s/60 sec at an angle of 120° at 0° for 20 h. The Salmonella serovar Braenderup H9812 standard served as size markers [41]. Specific experimental methods were performed as previously described [42].
Antimicrobial susceptibility testing
The isolates were subjected to antimicrobial susceptibility testing by the broth microdilution method, as described by Clinical and Laboratory Standards Institute (CLSI 2019) guidelines. E. coli ATCC® 25,922™ was used as a quality control strain. The results were interpreted with reference to CLSI 2019. For ENR and FFC, Veterinary Microbiology Laboratory Standards were applied [43].
Detection of major ARGs
Polymerase chain reaction (PCR) was used to detect the respective ARGs (Table S1) of chloramphenicol, macrolides, quinolones, β-lactams, tetracyclines, and aminoglycosides, which are widely used in animals. gyrA and parC targets in the quinolone resistance determining region (QRDR) of 7 strains with different PFGE types and high fluoroquinolone resistance [27] (MIC ≥ 32 mg/L) were detected. The gyrA and parC sequences were compared with those of E. coli K12 available in the NCBI database.
Detection of class I integron and variable region in fluoroquinolone-resistant E. Coli
A total of 7 with high fluoroquinolone resistance of different PFGE types integrase positive strains were detected by PCR. Identification of the variable region of class 1 integrons was detected in the case of integrase positive. The PCR products were recovered, ligated and transformed, and the plasmids were extracted. The plasmids were sent to Shanghai BioEngineering Co., Ltd. for sequencing. Results of the sequences in the integron were compared with those in the GenBank database (NCBI).
Plasmid transferability
The plasmid containing integron was used for plasmid transfer by conjugation or electroshock transformation. Salmonella H9812 was used as a recipient. The screening was performed using an SS medium containing 0.25 mg/L enrofloxacin. The results of conjugative plasmid transfer were determined by PCR amplification detection with Salmonella-specific primer JY27, plasmid extraction, and AMR spectrum changes, and calculated according to the formula:
where f is the conjugative transfer frequency, C is the zygote number, D is the donor number, X is the zygote dilution factor, and Y donor is the dilution factor.
Plasmids from multiple highly resistant E. coli were transferred into Stellar (E. coli HST08) competent cells by electroporation.
WGS
In order to analyze the gene-environment, the whole genome of E. coli C3 with high AMR and high transferability frequency was sequenced. Firstly, the genomic DNA was extracted, after which the purity, concentration and integrity were checked by Nanodrop, Qubit, and 0.35% agarose gel electrophoresis. Next, large fragments of DNA were recovered by the BluePippin automatic nucleic acid recovery system. After end repair, the DNA fragments were purified with magnetic beads to construct a Qubit library and sequenced using the Nanopore platform.
Reads were filtered for low-quality, adapters, and too short fragments (< 2000 bp in length) and applied to de novo assembly for error correction of assembled draft genomes. Canu v1.5 [44] software was used to assemble the filtered reads; Racon v3.4.3 software was used to correct the assembly results with third-generation reads, and Circlator v1.5.5 software to circulate and adjust the start site. Genome component analysis mainly included gene coding, non-coding RNA, gene island, CRISPR, and other analysis, while functional annotation mainly included general databases, such as nr, Uniprot, COG, KEGG and other databases, as well as CAZyme, PHI, CARD, and other proprietary databases annotation.
Statistical analysis
The phylogenetic tree model was constructed using MEGA6.0 based on the 16s rRNA sequences of the isolated strains. PFGE was analyzed using Bionumerics 8.0 software to characterize strain kinship. Snapgene software was used to conduct multiple alignment analyses of amino acid sequences, and the QRDR target mutations were obtained. The sequenced genes were processed using DNASTAR software.
Data Availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Abbreviations
- AMR:
-
Antimicrobial resistance
- MDR:
-
Multidrug-resistant
- WGS:
-
Whole-genome sequencing
- ARG:
-
Antibiotic resistance gene
- ARB:
-
Antibiotic resistant bacteria
- ICE:
-
Integrative and conjugative element
- PFGE:
-
Pulsed-field gel electrophoresis
- FFC:
-
Florfenicol
- TE:
-
Tetracycline
- CRO:
-
Ceftriaxone
- ENR:
-
Enrofloxacin
- MEM:
-
Meropenem
- PB:
-
Polymyxin B
- TGC:
-
Tigecycline
- MAR:
-
Marbofloxacin
- AZM:
-
Azaerythromycin
- CN:
-
Gentamicin
- CLSI:
-
Clinical and Laboratory Standards Institute
- PCR:
-
Polymerase chain reaction
- QRDR:
-
Quinolone resistance determining region
- MIC:
-
Minimum inhibitory concentration
References
Cui CY, Chen C, Liu BT, He Q, Wu XT, Sun RY, Zhang Y, Cui ZH, Guo WY, Jia Q. Co-occurrence of plasmid-mediated Tigecycline and Carbapenem Resistance in Acinetobacter spp. from Waterfowls and their neighboring environment. Antimicrob Agents Ch 2020, 64(5).
Laxminarayan R, Amábile-Cuevas CF, Cars O, Evans T, Røttingen J. UN High-Level Meeting on antimicrobials—what do we need? LANCET 2016, 388(10041):218–220.
Poirel L, Madec JY, Lupo A, Schink AK, Kieffer N, Nordmann P, Schwarz S. Antimicrobial Resistance in Escherichia coli. Microbiol Spectr 2018, 6(4).
Leotta GA, Deza N, Origlia J, Toma C, Chinen I, Miliwebsky E, Iyoda S, Sosa-Estani S, Rivas M. Detection and characterization of Shiga toxin-producing Escherichia coli in captive non-domestic mammals. Vet Microbiol. 2006;118(1–2):151–7.
Paraoan C, Rivera WL, Vital PG. Detection of class I and II integrons for the assessment of antibiotic and multidrug resistance among Escherichia coli isolates from agricultural irrigation waters in Bulacan, Philippines. J Environ Sci Heal B. 2017;52(5):306–13.
Jinfu LI, Ding H, Xiaoshen LI, Liu B, Jia SU, Yuan L, University HA. Detection and analysis of Escherichia coli Resistance Gene in recessive Infection of pigs in Henan Province. J Henan Agricultural Sci 2017.
Huddleston JR. Horizontal gene transfer in the human gastrointestinal tract: potential spread of antibiotic resistance genes. Infect Drug Resist. 2014;7:167–76.
Hampton T. Report reveals scope of US antibiotic resistance threat. Jama-J Am Med Assoc. 2013;310(16):1661–3.
Wu B, Qi Q, Zhang X, Cai Y, Yu G, Lv J, Gao L, Wei L, Chai T. Dissemination of Escherichia coli carrying plasmid-mediated quinolone resistance (PMQR) genes from swine farms to surroundings. Sci Total Environ. 2019;665:33–40.
Stokes HW, Hall RM. A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol Microbiol. 1989;3(12):1669–83.
Puvaca N, de Llanos FR. Antimicrobial Resistance in Escherichia coli strains isolated from humans and Pet animals. Antibiotics-Basel 2021, 10(1).
Mediavilla JR, Chen L, Mathema B, Kreiswirth BN. Global epidemiology of community-associated methicillin resistant Staphylococcus aureus (CA-MRSA). Curr Opin Microbiol. 2012;15(5):588–95.
Lutgring JD. Carbapenem-resistant Enterobacteriaceae: an emerging bacterial threat. Semin Diagn Pathol. 2019;36(3):182–6.
Ibrahim S, Al-Saryi N, Al-Kadmy I, Aziz SN. Multidrug-resistant Acinetobacter baumannii as an emerging concern in hospitals. Mol Biol Rep. 2021;48(10):6987–98.
He T, Wang R, Liu D, Walsh TR, Zhang R, Lv Y, Ke Y, Ji Q, Wei R, Liu Z, et al. Emergence of plasmid-mediated high-level tigecycline resistance genes in animals and humans. Nat Microbiol. 2019;4(9):1450–6.
Liu YY, Wang Y, Walsh TR, Yi LX, Zhang R, Spencer J, Doi Y, Tian G, Dong B, Huang X, et al. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis. 2016;16(2):161–8.
Xia R, Ren Y, Xu H. Identification of plasmid-mediated quinolone resistance qnr genes in multidrug-resistant Gram-negative bacteria from hospital wastewaters and receiving waters in the Jinan area, China. Microb Drug Resist. 2013;19(6):446–56.
Wozniak RA, Waldor MK. Integrative and conjugative elements: mosaic mobile genetic elements enabling dynamic lateral gene flow. Nat Rev Microbiol. 2010;8(8):552–63.
Pembroke JT, Mcgrath MM. The role of conjugative transposons in the Enterobacteriaceae. Cellular&Molecular Life Sciences 2002.
Coetzee JN, Datta N, Hedges RW. R factors from Proteus rettgeri. J Gen Microbiol. 1972;72(3):543–52.
Spagnoletti M, Ceccarelli D, Rieux A, Fondi M, Taviani E, Fani R, Colombo MM, Colwell RR, Balloux F. Acquisition and evolution of SXT-R391 integrative conjugative elements in the seventh-pandemic Vibrio cholerae lineage. MBIO 2014, 5(4).
Juhas M, Power PM, Harding RM, Ferguson DJ, Dimopoulou ID, Elamin AR, Mohd-Zain Z, Hood DW, Adegbola R, Erwin A, et al. Sequence and functional analyses of Haemophilus spp. genomic islands. Genome Biol. 2007;8(11):R237.
Sáenz Y, Briñas L, Domínguez E, Ruiz J, Torres C. Mechanisms of resistance in multiple-antibiotic-resistant Escherichia coli strains of Human, Animal, and Food origins. Antimicrob Agents Chemother. 2004;48(10):3996.
Bi D, Xu Z, Harrison EM, Tai C, Wei Y, He X, Jia S, Deng Z, Rajakumar K, Ou HY. ICEberg: a web-based resource for integrative and conjugative elements found in Bacteria. Nucleic Acids Res. 2012;40(Database issue):D621–6.
Collignon P, Powers JH, Chiller TM, Aidara-Kane A, Aarestrup FM. World Health Organization ranking of antimicrobials according to their importance in human medicine: a critical step for developing risk management strategies for the use of antimicrobials in food production animals. Clin Infect Dis. 2009;49(1):132–41.
Fadhil AI, Sabri AM. Plasmid-mediated mechanism of Quinolone Resistance on E. Coli isolates from different clinical samples. Arch Razi Inst. 2021;76(3):561–73.
Kariuki K, Diakhate MM, Musembi S, Tornberg-Belanger SN, Rwigi D, Mutuma T, Mutuku E, Tickell KD, Soge OO, Singa BO, et al. Plasmid-mediated quinolone resistance genes detected in Ciprofloxacin non-susceptible Escherichia coli and Klebsiella isolated from children under five years at hospital discharge, Kenya. BMC Microbiol. 2023;23(1):129.
Noel HR, Petrey JR, Palmer LD. Mobile genetic elements in Acinetobacter antibiotic-resistance acquisition and dissemination. Ann N Y Acad Sci. 2022;1518(1):166–82.
Ghaly TM, Gillings MR. New perspectives on mobile genetic elements: a paradigm shift for managing the antibiotic resistance crisis. Philos Trans R Soc Lond B Biol Sci. 2022;377(1842):20200462.
Singh M, Chaudhry MA, Yadava JN, Sanyal SC. The spectrum of antibiotic resistance in human and veterinary isolates of Escherichia coli collected from 1984-86 in northern India. J Antimicrob Chemoth. 1992;29(2):159–68.
Enne VI, Cassar C, Sprigings K, Woodward MJ, Bennett PM. A high prevalence of antimicrobial resistant Escherichia coli isolated from pigs and a low prevalence of antimicrobial resistant E. Coli from cattle and sheep in Great Britain at slaughter. Fems Microbiol Lett 2008, 278(2).
Makarov DA, Ivanova OE, Karabanov SY, Gergel MA, Pomazkova AV. Antimicrobial resistance of commensal Escherichia coli from food-producing animals in Russia. Vet World. 2020;13(10):2053–61.
Zhao S, Blickenstaff K, Bodeis-Jones S, Gaines SA, Tong E, McDermott PF. Comparison of the prevalences and antimicrobial resistances of Escherichia coli isolates from different retail meats in the United States, 2002 to 2008. Appl Environ Microb. 2012;78(6):1701–7.
Barlow RS, Pemberton JM, Desmarchelier PM, Gobius KS. Isolation and characterization of integron-containing bacteria without antibiotic selection. Antimicrob Agents Ch. 2004;48(3):838–42.
Rawat N, Anjali, Jamwal R, Devi PP, Yadav K, Kumar N, Rajagopal R. Detection of unprecedented level of antibiotic resistance and identification of antibiotic resistance factors, including QRDR mutations in Escherichia coli isolated from commercial chickens from North India. J Appl Microbiol. 2022;132(1):268–78.
Sunde M, Simonsen GS, Slettemeas JS, Bockerman I, Norstrom M. Integron, plasmid and Host Strain Characteristics of Escherichia coli from humans and food included in the Norwegian Antimicrobial Resistance Monitoring Programs. PLoS ONE. 2015;10(6):e128797.
Kaushik M, Khare N, Kumar S, Gulati P. High prevalence of Antibiotic Resistance and integrons in Escherichia coli isolated from Urban River Water, India. Microb Drug Resist. 2019;25(3):359–70.
Partridge SR, Kwong SM, Firth N, Jensen SO. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin Microbiol Rev 2018, 31(4).
Zadoks RN, Griffiths HM, Munoz MA, Ahlstrom C, Bennett GJ, Thomas E, Schukken YH. Sources of Klebsiella and Raoultella species on dairy farms: be careful where you walk. J Dairy Sci. 2011;94(2):1045–51.
Zhu Y, Liu W, Schwarz S, Wang C, Yang Q, Luan T, Wang L, Liu S, Zhang W. Characterization of a blaNDM-1-carrying IncHI5 plasmid from Enterobacter cloacae complex of food-producing animal origin. J Antimicrob Chemoth. 2020;75(5):1140–5.
Kang JY, Lee W, Noh GM, Jeong BH, Park I, Lee SJ. Fluoroquinolone resistance of Staphylococcus epidermidis isolated from healthy conjunctiva and analysis of their mutations in quinolone-resistance determining region. Antimicrob Resist In. 2020;9(1):177.
Xia LN, Li L, Wu CM, Liu YQ, Tao XQ, Dai L, Qi YH, Lu LM, Shen JZ. A survey of plasmid-mediated fluoroquinolone resistance genes from Escherichia coli isolates and their dissemination in Shandong, China. Foodborne Pathog Dis. 2010;7(2):207–15.
Watts JL. Performance standards for Antimicrobial disk and dilution susceptibility test for bacteria isolated from animals; approved standard: performance standards for antimicrobial disk and dilution susceptibilty tests for bacteria isolated from animals. approved standard; 2013.
Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–36.
Acknowledgements
Not applicable.
Funding
This work was supported by grants from the Jilin Natural Science Foundation (grant numbers YDJ202203CGZH050, 20210202033NC, 20220202060NC).
Author information
Authors and Affiliations
Contributions
L-C K, and H-X M contributed to the conception of the study. DQ, X-Y L and Y-Z Lperformed the data analyses and wrote the manuscript, D-M Z and G-M L ontributed significantly to analysis and manuscript preparation. P-H L and Y-L W helped in performing the analysis with constructive discussions. All authors contributed to the article have approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable (This experiment is not reported for living vertebrates.)
Consent for publication
Not applicable
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, D.m., Ding, Q., Li, P.h. et al. Antimicrobial resistance in E. Coli of animal origin and discovery of a novel ICE mobile element in Northeast China. BMC Vet Res 19, 255 (2023). https://doi.org/10.1186/s12917-023-03828-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-023-03828-5