
Abstract
Background
African swine fever (ASF) is an infectious transboundary animal disease which causes high mortality, approaching 100% in domestic pigs and it is currently considered as the most serious constraint to domestic pig industry and food security globally. Despite regular ASF outbreaks within Malawi, few studies have genetically characterized the causative ASF virus (ASFV). This study aimed at genetic characterization of ASFV responsible for the 2019 outbreak in northern Malawi. The disease confirmation was done by polymerase chain reaction (PCR) followed by molecular characterization of the causative ASFV by partial genome sequencing and phylogenetic reconstruction of the B646L (p72) gene, nucleotide alignment of the intergenic region (IGR) between I73R and I329L genes and translation of the central variable region (CVR) coded by B602L gene.
Results
All thirteen samples collected during this study in Karonga district in September 2019 were ASFV-positive and after partial genome sequencing and phylogenetic reconstruction of the B646L (p72) gene, the viruses clustered into ASFV p72 genotype II. The viruses characterized in this study lacked a GAATATATAG fragment between the I173R and the I329L genes and were classified as IGR I variants. Furthermore, the tetrameric amino acid repeats within the CVR of the B602L gene of the 2019 Malawian ASFV reported in this study had the signature BNDBNDBNAA, 100% similar to ASFV responsible for the 2013 and 2017 ASF outbreaks in Zambia and Tanzania, respectively.
Conclusions
The results of this study confirm an ASF outbreak in Karonga district in northern Malawi in September 2019. The virus was closely related to other p72 genotype II ASFV that caused outbreaks in neighboring eastern and southern African countries, emphasizing the possible regional transboundary transmission of this ASFV genotype. These findings call for a concerted regional and international effort to control the spread of ASF in order to improve nutritional and food security.
Similar content being viewed by others


Background
African swine fever (ASF) is a highly contagious, deadly hemorrhagic viral disease of domestic pigs and wild boars of all breeds and ages caused by ASF virus (ASFV), a double-stranded DNA arbovirus and the only member of the family Asfarviridae, genus Asfivirus [1,2,3]. The virus is transmitted through a sylvatic cycle involving warthogs (Phacochoerus africanus) which do not develop clinical disease and soft ticks of the Ornithodoros moubata complex inhabiting warthog burrows [4]. This ancient sylvatic cycle specific to eastern and southern Africa occasionally leads to virus spill to domestic pigs through the tick-pig cycle that involves infected soft ticks dropped by warthogs at pig shelters [5]. After the virus is introduced to the domestic pig population, transmission between domestic pig population occurs through the transmission of the virus among domestic pigs or by feeding contaminated pig products to domestic pigs, accounting for the majority of ASF outbreaks globally [6, 7]. Recently, an additional epidemiological cycle characterized by both direct transmission between infected and susceptible Eurasian wild boar (Sus scrofa) and indirect transmission through carcasses in the habitat has been described and named the wild boar-habitat cycle [8]. The current geographical distribution of ASF extends across more than 50 countries in Africa, Europe and Asia and among them, 33 are countries in Africa, south of Sahara [9, 10]. The recent spread of ASF within Europe and Asia has substantially increased the global concern regarding the disease and it is considered as the most devastating disease to global domestic pig industry and food security [2].
Depending on the isolate, the ASF viral genome varies in length from about 170 and 193 kilobase pairs (kbp) and encodes between 150 and 167 open reading frames with a conserved central region and variable termini [2]. Although most of the length variations were described to be associated with the gain or loss of copies within multigene families (MGF), smaller length variations are also associated with the number of tandem repeat sequences (TRS) located at loci both within coding and intergenic regions [11]. Sequence analysis of distinct genomic regions of ASFV has proved to be very useful in identifying the origin and transmission pathways of ASF during outbreaks [12]. Based on the ASFV p72 major capsid protein gene (B646L), 24 distinct ASFV genotypes (I-XXIV) have been described [13, 14] and analysis of additional genes has shown to provide higher resolution to distinguish between closely related isolates. The central variable region (CVR) within the B602L gene has shown to provide more information about relationship between isolates at genotype, country and regional levels [15,16,17,18]. Recent studies have demonstrated the value of the tetrameric repeat sequences (TRS) located in the intergenic region between the I73R and I329L genes in determining the origin and mapping the spread of closely related ASFV isolates [19, 20]. By combining p72, B602L (CVR) and TRS, a high level resolution is achieved for viral discrimination despite the existence of many other markers.
In Africa South of Sahara, the existence of all the 24 ASFV p72 genotypes described to date has been demonstrated [21, 22]. Briefly, in West Africa, where there is no evidence of the existence of the ASFV sylvatic cycle, only genotype I has been reported. In Central, Eastern and Southern Africa where three ASFV transmission cycles exist, rich ASFV genotypic variability exists with all 24 (I to XXIV) ASFV genotypes [9, 13, 22,23,24,25]. In Malawi, ASF is endemic and several outbreaks have been reported to the World Organization for Animal Health (OIE) in almost all its provinces [9, 10, 22, 26]. For instance, from January 2005 to December 2018, 227 ASF outbreaks which led to 87,063 pig deaths were reported to OIE [10]. Each year, ASF is reported in different parts of the country posing a serious constraint to the development of the domestic pig industry in Malawi. Despite the regular ASF outbreaks in domestic pigs within Malawi, molecular characterization of the causative viruses has been limited, thus the ASF outbreaks patterns and ASFV genotypes mapping in the country are incomplete. Most of ASFV molecular characterization studies from Malawi were carried out more than a decade ago and previously characterized ASFV strains grouped into p72 genotypes V, VIII and XII and all domestic and sylvatic ASFV transmission cycles have been described in the country [16, 18, 26, 27]. The tick-domestic pig and the sylvatic cycles of ASFV transmission involving warthogs and ticks collected from domestic pig shelters and warthogs’ burrows have been demonstrated in Malawi [28,29,30]. Warthogs and bush pigs which are natural reservoirs for ASFV are commonly found in National Parks and Wildlife Reserves of Malawi [28, 29, 31, 32] and may possibly play a role in the epidemiology of ASF in the country. Proper ASF outbreak investigation and continuous molecular characterization of the responsible viral strains provide insight into the transmission dynamics of the virus, differentiation of closely related strains and identification of potential transmission routes during and after outbreaks in order to guide appropriate interventions for an effective control of ASF [33]. This study aimed at confirming and conducting molecular characterization of the ASFV responsible for the 2019 outbreak in Karonga district located in northern Malawi.
Results
Laboratory confirmation of ASF
All collected tissue samples belonging to 13 different domestic pigs from Karonga district included in the present study were positive for ASFV after conducting diagnostic PCR using ASFV-specific primers.
Phylogenetic reconstruction of ASFV targeting B646L (p72) gene, TRS and CVR
In order to classify viruses characterized in this study among the 24 ASFV p72 known genotypes, the c-terminal end of B646L (p72) gene was amplified and sequenced. All sequences obtained in this study have been deposited to the GenBank and given accession numbers (Accession numbers MN755863-MN755874). The ASFV from domestic pigs in Karonga district named MAL/19/Karonga/1–4 had 100% nucleotide identity. The BLASTn of B646L (p72) nucleotide sequences of MAL/19/Karonga/1–4 against other ASFV strains available at GenBank showed 100% nucleotide identity with ASFV strains previously described in Tanzania, Zambia, Georgia, China, Vietnam, Estonia, Moldova, Czech Republic, Belgium and Poland. After phylogenetic reconstruction using ASFV strains indicated in Table 1, the MAL/19/Karonga/1–4 clustered together with ASFV belonging to genotype II (Fig. 1).

Maximum Likelihood phylogenetic reconstruction of African swine fever virus (ASFV) strains from Malawi together with genotype II ASFV previously described in Tanzania, Zambia, Mozambique, Zimbabwe, Georgia and China, sharing high nucleotide identity with viruses that caused outbreak in Malawi during September 2019 based on B646L (p72) gene nucleotide sequences. Phylogeny was inferred following 1000 bootstrap replications, and the node values show percentage bootstrap support. Scale bar indicates nucleotide substitutions per site. The square indicates the African swine fever virus strains that caused outbreak in Malawi during September 2019
The analysis of the intergenic region (IGR) between I73R and I329L genes of the strains that caused ASF outbreak in Karonga district in northern Malawi in 2019 showed 99.41% nucleotide identity with ASFV genotype II strains responsible for the 2017 outbreaks in Morogoro and Pwani regions of Tanzania and 99.16% nucleotide identity with some isolates circulating in Europe and Asia, including the Georgia 2007/1 isolate. The viruses characterized in this study lacked a GAATATATAG fragment between the I173R and the I329L genes (Fig. 2) and were classified as IGR I variants as previously described [19, 20]. In addition, a similar G to A replacement were observed in ASFV described in this study and those previously described in Tanzania (Fig. 2).

Nucleotide sequence alignment of the intergenic region between I73R and I329L genes of African swine fever virus strains belonging to B646L (P72) genotype II from Tanzania, Europe and China. The nucleotides highlighted in gray are absent in some viruses including the strains that caused outbreak in Malawi during September 2019. Also, a substitution of G by A is observed in Tanzanian and Malawian viruses only
The CVR sequences obtained in this study were translated into amino acids and coded to obtain corresponding signature. The CVR tetrameric repeats of ASFV that caused the outbreak in Karonga district in 2019 included CADT, NVDT, CASM, CAST and CSTS, corresponding to B, N, D and A codes, respectively. The ASFV characterized in this study showed 10 amino acid tetramers (BNDBNDBNAA) that were 100% identical to each other. A similarity search against other ASFV amino acids sequences performed by BLASTp showed 100% amino acids identity to ASFV that caused previous ASF outbreaks in Tanzania, Madagascar, Zambia, Mozambique, Mauritius, Russia and China (Table 2).
Discussion
In this study, we confirmed an ASF outbreak in Karonga district in northern Malawi that occurred during September 2019. Laboratory confirmation was done by PCR and subsequent genetic characterization of partial ASFV genome by phylogenetic reconstruction of the B646L (p72) gene, nucleotide alignment of the intergenic region (IGR) between I73R and I329L genes and amino acid alignment of the B602L (CVR) gene. After phylogenetic analysis, the ASFV strains obtained in this study clustered together with viruses belonging to ASFV p72 genotype II. Furthermore, the IGR and CVR signatures of ASFV strains obtained in this study showed high identity with p72 genotype II viruses previously described in Tanzania, Zambia, Mozambique, Zimbabwe, Georgia, China, Vietnam, Estonia, Moldova, Czech Republic, Belgium, Poland and Russia.
The nucleotide sequences of the c-terminal end of B646L (p72) gene of the viruses characterized in this study showed 100% nucleotide identity with those previously described in neighboring countries of Tanzania and Zambia. There is a high possibility of transboundary spread of ASFV between Malawi, Tanzania and Zambia since these countries share a common border and the towns of Karonga in Malawi, Kyela in Tanzania and Mbala in Zambia are less than 400 km apart. The ASFV strain that caused an outbreak in Kyela, Tanzania in 2010 had 100% nucleotide identity for the p72 (B646L) gene to Malawian ASFV collected from the 2011 outbreak in Karonga (Table 1). It was previously speculated that the introduction of ASFV genotype II, previously not described in Tanzania, occurred by importation of pig products from Karonga in Malawi [35, 36]. Since then, there has been persistent circulation of highly virulent genotype II viruses in the southern highlands of Tanzania, that have devastated nutritional and food security [40]. Since the introduction of genotype II viruses in Tanzania, these viruses have spread northwards within Tanzania causing devastating impact to the domestic pig industry and expanding the geographical range of this ASFV genotype [34, 39, 40]. Illegal transportation of infected pigs and pig products to uninfected areas have been cited to contribute to the spread of ASFV within Tanzania [34]. There is a need for higher control in order to prevent this ASFV genotype II from its northward spread otherwise it may reach other neighboring East African Community countries, such as Rwanda, Burundi, Uganda and Kenya. It is not uncommon for animal viruses to expand their geographical range as observed with peste des petits ruminants [41,42,43], Tilapia lake virus disease [44] and foot-and-mouth disease [45,46,47].
The analysis of the IGR between I73R and I329L genes showed high nucleotide identity with previously characterized ASFV genotype II strains and lacked a GAATATATAG fragment similar to ASFV strains circulating in Tanzania, different countries of Europe and Asia including the isolate Georgia 2007/1 collected in Georgia in 2007 that subsequently spread to other countries of eastern Europe and China [39, 48, 49]. In addition, we observed a similar G to A replacement in strains characterized in this study to ASFV strains responsible for the 2017 outbreaks in Morogoro and Pwani regions of Tanzania [39]. This suggest that ASFV genotype II strains circulating in Tanzania and Malawi are from probably the same source. Whole genome sequencing of strains described in this study and ASFV circulating in eastern and southern Africa will be able to discriminate closely related strains and establish more accurately epidemiological links between different ASF outbreaks occurring in the region.
The tetrameric amino acids repeats within the CVR of the B602L gene of the 2019 Malawian ASFV reported in this study had the signature BNDBNDBNAA which was 100% similar to the ASFV strains that caused the ASF outbreaks in Tanzania in 2017 [39] and during 2013 in Zambia [12]. The ZAM/13/Mbala virus was collected in April 2013 from domestic pigs reared in a village along the border with neighboring Tanzania and a suspected introduction from Tanzania through trans-border trade of pigs and pork products is speculated [12]. Kyela at the Tanzanian side, Karonga in northern Malawi and Mbala in Zambia share borders and ASF outbreaks have been reported to temporally coincide [12, 35, 36].
Comparison of the 3 ASFV genomic regions analyzed in this study revealed high identity between the strains characterized in this study and other viruses belonging to ASFV p72 genotype II that have caused previous outbreaks elsewhere including in Madagascar in 1998 [50]. The first ASFV reported in Madagascar was identical to the virus recovered from the 1994 ASF outbreak in Mozambique and was suspected to be the most likely source of ASF infection in Madagascar that was previously free from ASF [50, 51]. The ASFV p72 genotype II was recovered from domestic pigs in the year 2002 and from soft ticks in 2006 in Mozambique [14]. In 2007, the ASFV p72 genotype II strain with high identity to viruses previously described in Madagascar and Mozambique was reported in Mauritius for the first time and swill feeding to domestic pigs was suspected to be responsible for the introduction of the ASFV to the island [52]. The ASFV genotype II occurred in the Caucasus region of Georgia in 2007 with subsequent spread to Russia, different countries of Europe before it reached China in August 2018 and spread to neighboring Asian countries [19, 20, 49]. The virus responsible for the ASF outbreak in Georgia in 2007 was closely related to ASFV strains previously described in Mozambique, Zambia and Madagascar, thus the southern Africa countries or Madagascar were suspected to be the most likely source of ASF infection of the Georgia 2007 ASF outbreak [49]. Eastern and southern Africa countries including Malawi are characterized by the presence of wildlife protected areas with warthogs and ticks of the Ornithodoros moubata complex inhabiting warthogs’ burrows. These natural reservoir of the ASFV play an important role in the maintenance and transmission of the ASFV through the sylvatic cycle of the virus [14, 22, 53]. The 2015 ASF outbreak in Zimbabwe was caused by the ASFV genotype II after several years without ASF outbreak reported in the country and the transboundary spread from neighboring Mozambique was suspected [37]. The high identity between ASFV strains described in this study and viruses previously characterized in southern Africa countries, Madagascar, Europe and Asia suggests that they may have probably the same wild source and maintained through domestic cycle. In Malawi, the sylvatic cycle of ASFV involving ticks collected from warthogs habitat has been previously described [29] and a detailed study need to be carried out to assess the current role of wild suids and Ornithodoros ticks in the maintenance and transmission of ASFV in Malawi.
Conclusions
The virus responsible for the 2019 ASF outbreak in Karonga district clustered into p72 genotype II and showed high nucleotide identity with ASFV strains causing outbreaks in neighboring eastern and southern Africa countries suggesting that the same ASFV strains are causing outbreaks across borders. In addition, ASFV strains described in this study were closely related to viruses previously reported in Europe and Asia. These findings highlight the need for a concerted regional and international effort to control the spread of ASF in order to improve nutritional and food security. Investigation of the role of ASFV sylvatic cycle and further characterizations by whole genome sequencing are needed to fully understand molecular epidemiology of ASFV in Malawi.
Methods
Study area and sample collection
Samples used in this study were collected in Karonga district in northern Malawi from small-scale pig farmers in response to a report from local veterinarians to the Malawian National Veterinary Epidemiology Unit of a hemorrhagic disease affecting many domestic pigs with clinical symptoms suggestive of ASF in September 2019 (Fig. 3). Two tissue samples per domestic pig including spleen and liver were aseptically collected from thirteen dead pigs and transported to the laboratory. In the laboratory, samples were processed by homogenization in sterile phosphate-buffered saline (PBS) at a ratio of 1:10 w/v followed by centrifugation at 6000 g for 5 min and cryopreservation of the supernatant at − 80 ∘C until DNA extraction.

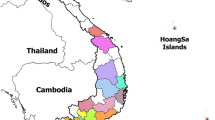
Distribution of African swine fever outbreaks reported in Malawi from 2016 to September 2019. Outbreaks occurred in all provinces of the country. Source of data: OIE World Animal Health Information System. The map was developed using QGIS version 3.4.4 (https://www.qgis.org/en/site/about/index.html)
DNA extraction and nucleotide amplification
QIAmp nucleic acid extraction kit (Qiagen, Hilden, Germany) was used for DNA extraction from collected samples, following manufacturer’s instructions. The presence of ASFV in collected samples was confirmed by polymerase chain reaction (PCR) using ASF diagnostic primers PPA1 and PPA2, as previously described by Aguero et al. [54]. The variable 3′-end of B646L gene encoding the major capsid protein p72, the tetramer amino acid repeats within the hypervariable central variable region (CVR) and the intergenic region (IGR) between I73R and I329L were amplified using the following primers: p72-D/p72-U [16], ORF9L-F/ORF9L-R [55] and ECO1A/ ECO1B [19], respectively.
Nucleotide sequencing and phylogenetic analysis
The nucleotide sequences of PCR products were obtained by automated dideoxynucleotide cycle sequencing using BigDye Terminator Cycle sequencing kit version 3.1 (Applied Biosystems, Foster City, CA). Sequence scanner software version 2.0 (Applied Biosystems, Foster City, CA) and Bioedit version 7.2.5 (Ibis Biosciences, Carlsbad, CA) were used to check the quality of raw sequences data and to obtain consensus nucleotide sequences from both forward and reverse primers for each of the amplified regions. The obtained consensus nucleotide sequences were used for BLASTn to search for similarity of nucleotide sequences obtained in this study to other nucleotide sequences available at GenBank. The dataset for p72 phylogenetic reconstruction consisted of 38 nucleotide sequences (402 characters), comprising of 4 sequences generated in this study and 34 homologous sequences from GenBank, including ASFV strains previously described in Malawi and genotype II ASFV strains from Tanzania, Zambia, Mozambique, Zimbabwe, Georgia and China, sharing high nucleotide identity with strains described in this study (Table 1). The phylogenetic tree construction was performed using Maximum Likelihood method and Kimura 2-parameter model with a bootstrap frequency of 1000 replicates as implemented by MEGA X [56]. The tandem repeat sequences (TRS) in the intergenic region between I73R and I329L genes of the strains characterized in this study were compared with other ASFV strains using CrustalW as implemented in MEGA X [56]. The central variable region nucleotide sequences of our ASFV isolates were translated and coded to obtain signatures based on previously reported codes [18, 34, 57]. A similarity search against other ASFV amino acid sequences was performed using BLASTp.
Availability of data and materials
The datasets generated and/or analysed during the current study are available at the GenBank repository (https//ncbi.nlm.nih.gov/genbank) with accession numbers MN755863 to MN755874.
Abbreviations
- ASF:
-
African swine fever
- ASFV:
-
African swine fever virus
- BLASTn:
-
Basic Local Alignment Search Tool for Nucleotides
- BLASTp:
-
Basic Local Alignment Search Tool for Proteins
- CVR:
-
Central variable region
- DNA:
-
Deoxyribonucleic acid
- IGR:
-
Intergenic region
- MEGA:
-
Molecular Evolutionary Genetics Analysis
- OIE:
-
World Organization for Animal Health
- P72:
-
African swine fever major capsid protein
- PBS:
-
Phosphate-buffered saline
- PCR:
-
Polymerase chain reaction
- TRS:
-
Tandem repeat sequences
References
Alonso C, Borca M, Dixon L, Revilla Y, Rodriguez F, Escribano JM, et al. ICTV virus taxonomy profile: Asfarviridae. J Gen Virol. 2018;99(5):613–4.
Dixon LK, Sun H, Roberts H. African swine fever. Antiviral Res. 2019;165:34–41.
Gilliaux G, Garigliany M, Licoppe A, Paternostre J, Lesenfants C, Linden A, et al. Newly emerged African swine fever virus strain Belgium/Etalle/wb/2018: complete genomic sequence and comparative analysis with reference p72 genotype II strains. Transbound Emerg Dis. 2019;66(6):2566–91.
Penrith M-L. History of “swine fever” in southern Africa. J S Afr Vet Assoc. 2013;84(1):1–6.
Ravaomanana J, Michaud V, Jori F, Andriatsimahavandy A, Roger F, Albina E, et al. First detection of African swine fever virus in Ornithodoros porcinus in Madagascar and new insights into tick distribution and taxonomy. Parasit Vectors. 2010;3(1):115.
Chenais E, Depner K, Guberti V, Dietze K, Viltrop A, Ståhl K. Epidemiological considerations on African swine fever in Europe 2014–2018. Porc Health Manag. 2019;5(1):6.
Penrith ML, Vosloo W. Review of African swine fever: transmission, spread and control. J S Afr Vet Assoc. 2009;80(2):58–62.
Chenais E, Ståhl K, Guberti V, Depner K. Identification of wild boar–habitat epidemiologic cycle in African swine fever epizootic. Emerg Infect Dis J- CDC. 2018;24:4 [cited 2019 Nov 11]; Available from: https://wwwnc.cdc.gov/eid/article/24/4/17-2127_article.
Mfumu LKM, Saegerman C, Dixon LK, Madimba KC, Kazadi E, Mukalakata NT, et al. African swine fever: update on eastern, central and southern Africa. Transbound Emerg Dis. 2019; [cited 2019 Mar 31]; Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/tbed.13187.
OIE. World Animal Health Information System [Internet]. 2020 [cited 2020 Apr 28]. Available from: http://www.oie.int/wahis_2/public/wahid.php/Countryinformation/Reporting.
Giammarioli M, Gallardo C, Oggiano A, Iscaro C, Nieto R, Pellegrini C, et al. Genetic characterisation of African swine fever viruses from recent and historical outbreaks in Sardinia (1978–2009). Virus Genes. 2011;42(3):377–87.
Simulundu E, Chambaro HM, Sinkala Y, Kajihara M, Ogawa H, Mori A, et al. Co-circulation of multiple genotypes of African swine fever viruses among domestic pigs in Zambia (2013–2015). Transbound Emerg Dis. 2017;65(1):114–22.
Achenbach JE, Gallardo C, Nieto-Pelegrín E, Rivera-Arroyo B, Degefa-Negi T, Arias M, et al. Identification of a new genotype of African swine fever virus in domestic pigs from Ethiopia. Transbound Emerg Dis. 2017;64(5):1393–404.
Quembo CJ, Jori F, Vosloo W, Heath L. Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound Emerg Dis. 2018;65(2):420–31.
Atuhaire DK, Afayoa M, Ochwo S, Mwesigwa S, Okuni JB, Olaho-Mukani W, et al. Molecular characterization and phylogenetic study of African swine fever virus isolates from recent outbreaks in Uganda (2010–2013). Virol J. 2013;10(1):247.
Bastos ADS, Penrith M-L, Crucière C, Edrich JL, Hutchings G, Roger F, et al. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch Virol. 2003;148(4):693–706.
Gallardo C, Okoth E, Pelayo V, Anchuelo R, Martin E, Simon A, et al. African swine fever viruses with two different genotypes, both of which occur in domestic pigs, are associated with ticks and adult warthogs, respectively, at a single geographical site. J Gen Virol. 2011;92(2):432–44.
Nix RJ, Gallardo C, Hutchings G, Blanco E, Dixon LK. Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Arch Virol. 2006;151(12):2475–94.
Gallardo C, Fernández-Pinero J, Pelayo V, Gazaev I, Markowska-Daniel I, Pridotkas G, et al. Genetic variation among African swine fever genotype II viruses, eastern and Central Europe. Emerg Infect Dis. 2014;20(9):1544.
Ge S, Li J, Fan X, Liu F, Li L, Wang Q, et al. Molecular characterization of African swine fever virus, China, 2018. Emerg Infect Dis. 2018;24(11):2131–3.
Mulumba-Mfumu LK, Saegerman C, Dixon LK, Madimba KC, Kazadi E, Mukalakata NT, et al. African swine fever: update on eastern, central and southern Africa. Transbound Emerg Dis. 2019;66(4):1462–80.
Penrith M-L, Bastos AD, Etter EMC, Beltrán-Alcrudo D. Epidemiology of African swine fever in Africa today: sylvatic cycle versus socio-economic imperatives. Transbound Emerg Dis. 2019;66(2):672–86.
Couacy-Hymann E, Kouakou KV, Achenbach JE, Kouadio L, Koffi YM, Godji HP, et al. Re-emergence of genotype I of African swine fever virus in Ivory Coast. Transbound Emerg Dis. 2019;66(2):882–96.
Molini U, Mushonga B, Settypalli TBK, Dundon WG, Khaiseb S, Jago M, et al. Molecular characterization of African swine fever virus from outbreaks in Namibia in 2018. Transbound Emerg Dis. 2019;0(ja) [cited 2019 Oct 28]. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/tbed.13399.
Wade A, Achenbach JE, Gallardo C, Settypalli TBK, Souley A, Djonwe G, et al. Genetic characterization of African swine fever virus in Cameroon, 2010-2018. J Microbiol Seoul Korea. 2019;57(4):316–24.
Haresnape JM, Lungu SA, Mamu FD. An updated survey of African swine fever in Malawi. Epidemiol Infect. 1987;99(3):723–32.
Lubisi BA, Bastos ADS, Dwarka RM, Vosloo W. Molecular epidemiology of African swine fever in East Africa. Arch Virol. 2005;150(12):2439–52.
Haresnape JM, Lungu SA, Mamu FD. A four-year survey of African swine fever in Malawi. J Hyg (Lond). 1985;95(2):309–23.
Haresnape JM, Wilkinson PJ, Mellor PS. Isolation of African swine fever virus from ticks of the Ornithodoros moubata complex (Ixodoidea: Argasidae) collected within the African swine fever enzootic area of Malawi. Epidemiol Infect. 1988;101(1):173–85.
Haresnape JM, Wilkinson PJ. A study of African swine fever virus infected ticks (Ornithodoros moubata) collected from three villages in the ASF enzootic area of Malawi following an outbreak of the disease in domestic pigs. Epidemiol Infect. 1989;102(3):507–22.
Briggs P, Bartlett M-A. Malawi: the Bradt travel guide. Bradt travel guides; 2006. p. 276.
Morris B. Wildlife and landscapes in Malawi: selected essays on natural history. Victoria, BC, Canada: Trafford Publishing; 2009. p. 328.
Malogolovkin A, Burmakina G, Titov I, Sereda A, Gogin A, Baryshnikova E, et al. Comparative analysis of African swine fever virus genotypes and serogroups. Emerg Infect Dis. 2015;21(2):312–5.
Misinzo G, Kwavi DE, Sikombe CD, Makange M, Peter E, Muhairwa AP, et al. Molecular characterization of African swine fever virus from domestic pigs in northern Tanzania during an outbreak in 2013. Tropl Anim Health Prod. 2014;46(7):1199–207.
Misinzo G, Kasanga CJ, Mpelumbe Ngeleja C, Masambu J, Kitambi A, Van Doorsselaere J. African swine fever virus, Tanzania, 2010–2012. Emerg Infect Dis. 2012;18(12):2081–3.
Simulundu E, Sinkala Y, Chambaro HM, Chinyemba A, Banda F, Mooya LE, et al. Genetic characterisation of African swine fever virus from 2017 outbreaks in Zambia: identification of p72 genotype II variants in domestic pigs. Onderstepoort J Vet Res. 2018;85(1):5.
van Heerden J, Malan K, Gadaga BM, Spargo RM. Reemergence of African swine fever in Zimbabwe, 2015. Emerg Infect Dis. 2017;23(5):860–1.
Chapman DAG, Darby AC, Da Silva M, Upton C, Radford AD, Dixon LK. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg Infect Dis. 2011;17(4):599–605.
Chang’a JS, Mayenga C, Settypalli TBK, Achenbach JE, Mwanandota JJ, Magidanga B, et al. Symptomatic and asymptomatic cases of African swine fever in Tanzania. Transbound Emerg Dis. 2019;66(6):2402–10.
Fasina FO, Kissinga H, Mlowe F, Mshang’a S, Matogo B, Mrema A, et al. Drivers, risk factors and dynamics of African swine fever outbreaks, southern highlands, Tanzania. Pathogens. 2020;9(3):155.
Misinzo G, Kgotlele T, Muse EA, Doorsselaere JV, Berg M, Munir M. Peste des Petits ruminants virus lineage II and IV from goats in southern Tanzania during an outbreak in 2011. Br J Virol. 2015;2(1):1–4.
Shahriari R, Khodakaram-Tafti A, Mohammadi A. Molecular characterization of Peste des Petits ruminants virus isolated from four outbreaks occurred in southern Iran. BMC Vet Res. 2019;15(1):177.
Mugimba KK, Chengula AA, Wamala S, et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. J Fish Dis. 2018;00:1–9. https://doi.org/10.1111/jfd.12790.
Mugimba KK, Chengula AA, Wamala S, Mwega ED, Kasanga CJ, Byarugaba DK, et al. Detection of tilapia lake virus (TiLV) infection by PCR in farmed and wild Nile tilapia (Oreochromis niloticus) from Lake Victoria. J Fish Dis. 2018;00:1–9. https://doi.org/10.1111/jfd.12790.
Klein J, Parlak Ü, Özyörük F, Christensen LS. The molecular epidemiology of foot-and-mouth disease virus serotypes a and O from 1998 to 2004 in Turkey. BMC Vet Res. 2006;2(1):35.
Le VP, Vu TTH, Duong H-Q, Than VT, Song D. Evolutionary phylodynamics of foot-and-mouth disease virus serotypes O and a circulating in Vietnam. BMC Vet Res. 2016;12(1):269.
Tekleghiorghis T, Moormann RJM, Weerdmeester K, Dekker A. Foot-and-mouth disease transmission in Africa: implications for control, a review. Transbound Emerg Dis. 2016;63(2):136–51.
Li L, Ren Z, Wang Q, Ge S, Liu Y, Liu C, et al. Infection of African swine fever in wild boar, China, 2018. Transbound Emerg Dis. 2019;66(3):1395–8.
Rowlands RJ, Michaud V, Heath L, Hutchings G, Oura C, Vosloo W, et al. African swine fever virus isolate, Georgia, 2007. Emerg Infect Dis. 2008;14(12):1870–4.
Bastos ADS, Penrith M-L, Macome F, Pinto F, Thomson GR. Co-circulation of two genetically distinct viruses in an outbreak of African swine fever in Mozambique: no evidence for individual co-infection. Vet Microbiol. 2004;103(3–4):169–82.
Gonzague M, Roger F, Bastos A, Burger C, Randriamparany T, Smondack S, et al. Isolation of a non-haemadsorbing, non-cytopathic strain of African swine fever virus in Madagascar. Epidemiol Infect. 2001;126(3):453–9.
Lubisi BA, Dwarka RM, Meenowa D, Jaumally R. An investigation into the first outbreak of African swine fever in the Republic of Mauritius. Transbound Emerg Dis. 2009;56(5):178–88.
Jori F, Vial L, Penrith ML, Pérez-Sánchez R, Etter E, Albina E, et al. Review of the sylvatic cycle of African swine fever in sub-Saharan Africa and the Indian ocean. Virus Res. 2013;173(1):212–27.
Agüero M, Fernández J, Romero L, Sánchez Mascaraque C, Arias M, Sánchez-Vizcaíno JM. Highly sensitive PCR assay for routine diagnosis of African swine fever virus in clinical samples. J Clin Microbiol. 2003;41(9):4431–4.
Irusta PM, Borca MV, Kutish GF, Lu Z, Caler E, Carrillo C, et al. Amino acid tandem repeats within a late viral gene define the central variable region of African swine fever virus. Virology. 1996;220(1):20–7.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–9.
Boshoff CI, Bastos ADS, Gerber LJ, Vosloo W. Genetic characterisation of African swine fever viruses from outbreaks in southern Africa (1973–1999). Vet Microbiol. 2007;121(1):45–55.
Acknowledgements
The authors are especially grateful to Mariam R. Makange, Mhoja Ndalahwa and Anna Rogath Masawe for their excellent technical assistance during the laboratory analysis.
Funding
The study was funded by the Partnership for Skills in Applied Sciences, Engineering and Technology (PASET) through the Regional Scholarship and Innovation Fund (RSIF) awarded to JNH to carry out doctoral studies at SACIDS Africa Centre of Excellence for Infectious Diseases, SACIDS Foundation for One Health, Sokoine University of Agriculture, Morogoro, Tanzania. The funder had no role in study design, data collection and analysis, decision to publish as well as in the preparation of this manuscript. The findings and conclusions of this study are those of the authors and do not necessarily represent the views of RSIF-PASET.
Author information
Authors and Affiliations
Contributions
JNH participated in sample collection, laboratory analysis, data analysis and development of the first draft of the manuscript. GK participated in the study design, sample collection and actively revised the manuscript. GM and JNH conceived the idea, analyzed, interpreted data and revised the manuscript. OK, GM, HJN and JLCC contributed to the conception of the idea, design, interpretation of data and actively revised the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The samples used in this study were collected from naturally dead domestic pigs according to common veterinary practice as part of routine veterinary investigation by the District Veterinary Officer under the supervision of the national veterinary epidemiologist according to the Malawian Control and Diseases of Animals Act (CAP 66:02 of 1967) and to rule 6 of the Swine Fever Rules G.N. 209/1968. Oral consent was obtained from the domestic pig owners before sampling of their dead domestic pigs and documented in the District Veterinary Officer registry.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hakizimana, J.N., Kamwendo, G., Chulu, J.L.C. et al. Genetic profile of African swine fever virus responsible for the 2019 outbreak in northern Malawi. BMC Vet Res 16, 316 (2020). https://doi.org/10.1186/s12917-020-02536-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-020-02536-8