
Abstract
Background
Johne’s disease (JD) is a chronic intestinal inflammatory disease caused by Mycobacterium avium subsp. paratuberculosis (MAP) infection in ruminants. Since there are currently no effective vaccine or treatment options available to control JD, genetic selection may be an alternative strategy to enhance JD resistance. Numerous Single Nucleotide Polymorphisms (SNPs) have been reported to be associated with MAP infection status based on published genome-wide association and candidate gene studies. The main objective of this study was to validate these SNPs that were previously identified to be associated with JD by testing their effect on Holstein bulls’ estimated breeding values (EBVs) for milk ELISA test scores, an indirect indicator of MAP infection status in cattle.
Results
Three SNPs, rs41810662, rs41617133 and rs110225854, located on Bos taurus autosomes (BTA) 16, 23 and 26, respectively, were confirmed as significantly associated with Holstein bulls’ EBVs for milk ELISA test score (FDR < 0.01) based on General Quasi Likelihood Scoring analysis (GQLS) analysis. Single-SNP regression analysis identified four SNPs that were associated with sire EBVs (FDR < 0.05). This includes two SNPs that were common with GQLS (rs41810662 and rs41617133), with the other two SNPs being rs110494981 and rs136182707, located on BTA9 and BTA16, respectively.
Conclusions
The findings of this study validate the association of SNPs with JD MAP infection status and highlight the need to further investigate the genomic regions harboring these SNPs.
Similar content being viewed by others

Background
Johne’s disease (JD), also known as paratuberculosis, manifests as chronic enteritis in cattle and is caused by infection with the Gram-positive bacteria Mycobacterium avium subsp. paratuberculosis (MAP). With its worldwide prevalence, the disease is responsible for significant economic losses to the dairy industry [1]. Albeit controversial and debatable, JD etiological agent MAP is also viewed as a pathogen with zoonotic effects. Report of isolation of MAP from intestines of human patients suffering from Crohn’s disease has raised public health concerns [2]. Factors like non-availability of an efficacious vaccine to combat MAP infection, issues associated with currently available JD diagnostic assays such as long turnaround time associated with MAP culture tests, low sensitivity of ELISA tests during early stages of JD, and absence of efficient treatment options have limited JD control around the world [3]. Heritability estimates using different phenotypes of JD have been reported and are found to range from 0.06 to 0.27 [4,5,6,7] suggesting that there is enough genetic variation to enable selection for reduced susceptibility to MAP infection [8]. Given the low-to-moderate heritability estimates of JD resistance, the difficulty to collect accurate phenotypes on a large number of animals, and the fact that that animals are tested for MAP infection at a later life stage (i.e., do not have own record at selection stage), the use of genomic information is a promising way to make genetic progress for JD resistance.
Genome-wide association (GWAS) and candidate gene studies concerning JD have identified numerous single nucleotide polymorphisms (SNPs) across the bovine genome which are significantly associated with JD status in dairy cattle [9,10,11,12,13,14,15,16,17]. The identification of JD associated SNPs and the heritable nature of MAP infection reflect the genetic variation in susceptibility and resistance to JD. These identified SNPs could be used in JD resistance breeding programs based on marker-enhanced selection (MES) once they are included in genotyping platforms. However, before this can occur there is a need to validate them, especially using independent cattle populations.
In a recent study by Brito et al. [18], the authors reported genetic parameters such as heritability for MAP-specific antibody response and estimated breeding values (EBVs) for Holstein cattle based on milk ELISA test records along with their correlation with other economically important trait like milk yield; and other routinely evaluated traits such as somatic cell score (SCS); reproduction traits (calving to first service, 56-d non-return rate, Number of services, cows, First service to conception, cows, Days open; longevity trait (Direct herd life); and confirmation traits (Overall feet and legs, Overall conformation). Milk ELISA is a JD diagnostic method that detects MAP-specific antibodies in animals exposed to MAP and is therefore an indirect indicator of MAP infection status in cattle [19]. Unlike direct diagnostic tests based on MAP culture tests, MAP ELISA test have a quick turnaround time and can be easily used at the herd level. As studies concerning validation of JD SNPs are lacking, sires with highly accurate EBVs for milk ELISA testing can be used to validate previously identified JD SNPs. Therefore, the main objective of this study was to validate some of the previously associated JD SNPs in literature by testing their association with sire EBVs for milk ELISA test score.
Results
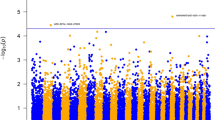
A total of 141 SNPs passed the quality control test for MAF threshold and were included in the association analyses. After the General Quasi Likelihood Scoring (GQLS) association analysis, the SNPs rs41810662 (P-value = 0.00011), rs41617133 (P-value = 4.5E-06) and rs110225854 (P-value = 6.4E-19) were found to be significantly associated with Holstein sire EBVs for milk ELISA test scores at a FDR of 1%, and no other SNPs were found to be significant at a FDR of 5%. Table 1 lists the significant SNPs based on GQLS analysis. The Manhattan plot of GQLS analysis is shown in Fig. 1. A total of four SNPs were found to be significantly associated with sire EBVs by single-SNP regression analysis. This included the SNPs rs41810662 (P-value = 0.00062) and rs41617133 (P-value = 0.00050) that were also identified in the GQLS analysis; and two other SNPs, rs110494981 (P-value = 9.0E-05) and rs136182707 (P-value = 0.00088), which are located on BTA9 and BTA16, respectively. Table 2 lists all the significant SNPs along with their estimated SNP effect based on single-SNP regression analysis. The Manhattan plot of single SNP regression analysis is shown in Fig. 2.

Manhattan plot for General Quasi Likelihood Score association analysis. A total of 3 SNPs across 3 chromosomes (BTA16, BTA23 and BTA26) were found to be significant at FDR < 0.01

Manhattan plot for Single SNP regression analysis. A total of 4 SNPs across 3 chromosomes (BTA9, BTA16 and BTA23) were found to be significant at FDR < 0.05
Discussion
With a global herd level prevalence ranging between 7 and 60% [20], Johne’s disease is a severe production limiting disease with significant animal welfare concerns to the worldwide dairy industry. Annual production losses due to JD on US dairy industry alone is estimated to be to $200–$250 millions dollars [1]. The losses associated with JD are mainly due to reduction in milk production, premature culling of JD positive animals, the management costs associated with JD control programs to limit MAP spread within and across the herds [21]. The role of host genetics in influencing JD infection status in cattle has been extensively studied, which resulted in the identification of several impactful genetic markers across the cattle genome [18]. However, studies concerning validation of genetic markers are still lacking. In this study, we validated some SNPs previously associated with JD by testing their association with breeding values estimated for JD milk ELISA test scores. A total of 498 bulls were classified into high (n = 248) and low (n = 250) groups based on their EBVs for milk ELISA test scores. Both groups were then genotyped using a customized SNP panel comprising 155 of the most prominent JD SNPs reported in the literature, including SNP from studies that used phenotypes other than milk ELISA test to define the case-control populations. Two association analyses were used to carry out SNP validation: a) General Quasi Likelihood Scoring (GQLS) analysis based on logistic regression, and, b) single-SNP regression.
Three SNPs rs41810662, rs41617133 and rs110225854 were found to be significantly associated with Holstein sire EBVs using GQLS analysis at 1% FDR. The association of these three SNPs with sire EBVs in the current study confirmed their association with JD. The SNP rs41810662 is located on BTA16 and was previously found to be associated with MAP antibody response in Holstein cows [22]. The other two SNPs (rs41617133 and rs110225854) are located on BTA23 and BTA26, respectively, and were previously reported to be associated with JD susceptibility in Holsteins [14]. These last two SNPs were selected from a GWAS study that used fecal culture along with blood ELISA to define MAP infection status. This may be indicative of the shared genomic region in Holstein cattle that influences both JD fecal culture and ELISA positivity and a stronger evidence of these SNPs’ role towards resistance to MAP infection.
A total of four SNPs were found to be significantly associated with sire EBVs by single-SNP regression analysis at 5% FDR. This included the SNPs rs41810662 and rs41617133 that were also identified in the GQLS analysis; and two other SNPs, rs110494981 and rs136182707, which are located on BTA9 and BTA16, respectively. The SNP rs110494981 was previously found to be associated with serum ELISA positivity for MAP antibodies in Italian Holsteins [13], whereas rs136182707 was previously identified by Mallikarjunappa et al. [22] and was found to be associated with MAP antibody ELISA positivity.
One main result from this study is that most of the SNPs (137 out of 141) were not found to be significant in the population under study. This is not uncommon and may have multiple causes. While several GWAS studies have enabled identification of many SNPs associated with MAP infection status in cattle, there seems to be little congruence among them, which confirms the complex polygenic nature of the disease [8, 18]. Among other factors, the choice of phenotype (e.g., milk or blood ELISA, fecal MAP culture, tissue MAP culture) used to define infection status in case-control studies is shown to impact results [23]. Each test differs in their specificity and sensitivity in accurately diagnosing MAP infection and results are often dictated by the stage of disease progression in the tested animal [24]. As a result, precise phenotyping of infected versus non-infected animals remains a challenge.
Milk ELISA is an indirect test for diagnosing MAP infection status that is based on identification of MAP-specific antibodies in MAP exposed animals. Unlike MAP culture techniques that take weeks to diagnose MAP infection, ELISA tests are more feasible at the herd level because of their rapid turn-around time and measurement cost. The objective of this study was to validate the previously reported JD SNPs reported in the literature. For this, we utilized sire EBVs estimated for milk ELISA test score to validate JD SNPs and confirm their association. As the sire EBVs used here were estimated using a large dataset of milk ELISA records, testing the association of JD SNPs with sire EBVs allows their validation at a large population level, while reducing the costs associated with genotyping large number of daughters. Genome-wide studies using EBVs to estimate SNP effects have been previously reported [25]. We adapted selective genotyping approach in our study where sires with EBVs estimated for milk ELISA results were classified into high and low groups and were subsequently genotyped. This approach of genotyping animals with extreme phenotypes reduces the need to genotype large number of animals and also increases power in determining the association of variants/SNPs in customized genotyping study like ours [26, 27].
Only few SNPs were validated while using EBVs estimated for milk ELISA test results. The specificity of milk ELISA test is high (99%); however, it lacks sensitivity during early stages of MAP infection [28]. This could perhaps have influenced the results of this study. Kirkpatrick et al. [8] reported that combining fecal culture and ELISA tests is more suitable in defining MAP infection status. Future studies could aim at determining EBVs for both tests combined to validate other JD SNPs. Two SNPs (rs41810662 and rs136182707) that were validated in this study were previously reported in a GWAS we conducted with Canadian Holstein cows for which no pedigree information was available [22]. Since no pedigree information was available for these Canadian Holsteins cows, their relatedness with the Canadian sires used in the current study could not be explained.
To the best of our knowledge, this is one of the first studies that considered validation of SNPs associated with JD reported in the literature using sire EBVs for milk ELISA test score. A follow-up study will consider search for gene variants in the same chromosomal areas as the identified SNP, which could play a role in JD resistance. One of the limitations associated with the current study is that validation was limited to a relatively small number of selected JD SNPs from GWAS and candidate gene studies from the literature. Future studies could consider validation of a larger number of reported JD SNPs using different (and ideally independent) dairy cattle populations.
The problems related with JD control, and the economic and welfare implications of the disease on the dairy industry, warrant extensive exploration of genetic selection as an alternative option to control JD. This begins with the validation of associated genetic markers. Validation of some of the SNPs in this study is a step in this direction and has prompted future consideration of similar validation studies using different phenotypes. It is a critical step before genetic marker-based selection can be implemented to breed for JD resistance in cattle.
Conclusions
Using sire EBVs estimated for milk ELISA test scores, a total of four previously reported JD related SNPs were validated in this study which can be used to optimize genomic selection schemes. Future studies will consider exploring the genomic regions surrounding the validated SNPs for the presence of any candidate genes and genetic variants with relevance to JD.
Methods
Resource population, phenotype classification and genotyping
The EBVs for Holstein sires were calculated for milk ELISA test score as described earlier by Brito et al. [18]. These EBVs were estimated using univariate linear animal models using different data subsets in 3 different scenarios: a) the complete data set (all herds); b) herds with at least one suspect or test-positive animal (ELISA optical density ≥ 0.07); and c) SCEN3: herds with at least one test-positive animal (ELISA optical density ≥ 0.11). Out of 5285 sires with EBVs estimated for milk ELISA test score, a total of 498 were classified into high (n = 248) and low (n = 250) EBV groups for selective genotyping. The EBVs of the selected sires from both the groups ranged from − 0.0677 to 0.0275 with a standard deviation of 0.0151. All sires had at least 30 daughters. Genotyping was performed using a customized SNP panel comprised of 155 of the most prominent JD SNPs reported in the literature, which included SNP from studies that used phenotypes other than milk ELISA to define their case-control populations (such as serum ELISA, tissue and fecal MAP culture) and SNPs from candidate-gene studies [[9, 10, 12,13,14, 17, 22, 29,30,31,32,33,34,35,36,37]; Supplementary File A]. These SNPs were located on all Bos taurus autosomes (BTA) except BTA 24. A pedigree file containing 7479 animals was generated by tracing back the pedigrees of sires with data up to 18 generations ago.
Quality control and statistical analyses
Quality control was applied to remove SNPs with minor allele frequency (MAF) less than 0.01 (or 1%). The association analysis was carried out using the Generalized Quasi-Likelihood Score method (GQLS) [38], implemented in the SNP1101 software [31]. The GQLS method is based on logistic regression and involves regression of EBVs versus SNP genotypes which are considered as response variables [38]. It further accounts for population substructure, by adjusting for relatedness among selected animals based on the pedigree-based relationship coefficients [39], and is not biased for selective genotyping. The GQLS statistical model can be defined as:
Where μi is the expected SNP allele frequency; Xi is the pseudo-phenotype (sire EBVs); Yi is the genotype of the SNP, considering Yi = 1/2 * (genotype code for the ith animal). The genotypes were coded as “0”, “1”, and “2” based on the number of reference alleles, corresponding to the respective proportions of 0, 1/2 and 1; β0 is a constant and β1 is the slope coefficient. In order to verify the association between each marker and the trait (sire EBV for milk ELISA test score), the null hypothesis was: H0: β1 = 0, i.e. the marker is not associated with the trait; while the alternate hypothesis was: H1: β1 ≠ 0, i.e. the marker is associated with the trait. To account for testing of multiple comparisons and identify significant SNPs associated with sire EBVs, genome-wise false discovery rates (FDR) of 1 and 5% were applied. In addition to GQLS analysis, single-SNP regression analysis including the additive polygenic effect was carried out. Unlike GQLS analysis, single-SNP regression allows for accounting for polygenic effect. The single-SNP regression model can be defined as:
where y is a vector with the sires’ EBV for milk ELISA test scores; μ is the overall mean value of the EBVs; b is the additive allele substitution effect (linear regression coefficient) of a SNP; x is the vector of number of copies of a given SNP allele (coded as 0, 1, or 2 for BB, AB, and AA, respectively); Z is the incidence matrix linking additive polygenic effects to bull EBVs; a is the vector of additive polygenic effects; and e is a vector of the residual effects. The model assumptions are as follows: a follows a normal distribution N (0, Gσ2a), in which G is the genomic relationship matrix [40], and σ2a is the additive genetic variance.
Availability of data and materials
The sire EBVs were provided by Brito et al. 2018 [18] and were stored in the Centre for Genetic Improvement of Livestock (CGIL) repository (http://cgil.uoguelph.ca/). These datasets are not publicly available due to on-going analyses but are available from the senior author (NK) on reasonable request.
Abbreviations
- JD:
-
Johne’s disease
- MAP:
-
Mycobacterium avium subsp. paratuberculosis
- SNPs:
-
Single nucleotide polymorphisms
- EBVs:
-
Estimated breeding values
- GQLS:
-
General quasi likelihood scoring
- MES:
-
Marker-enhanced genetic selection
- MAF:
-
Minor allele frequency
References
Ott SL, Wells SJ, Wagner BA. Herd-level economic losses associated with Johne’s disease on US dairy operations. Prev Vet Medicine. 1999;40:179–92.
Scanu AM, Bull TJ, Cannas S, Sanderson JD, Sechi LA, Dettori G, et al. Mycobacterium avium subspecies paratuberculosis infection in cases of irritable bowel syndrome and comparison with Crohn’s disease and Johne’s disease: common neural and immune pathogenicities. J Clin Microbiol. 2007;45:3883–90.
Mallikarjunappa S, Adnane M, Cormican P, Karrow NA, Meade KG. Characterization of the bovine salivary gland transcriptome associated with Mycobacterium avium subsp paratuberculosis experimental challenge. BMC Genomics. 2019;20:491.
Mortensen H, Nielsen SS, Berg P. Genetic variation and heritability of the antibody response to Mycobacterium avium subspecies paratuberculosis in Danish Holstein cows. J Dairy Sci. 2004;87:2108–13.
Gonda MG, Chang YM, Shook GE, Collins MT, Kirkpatrick BW. Genetic variation of Mycobacterium avium ssp. paratuberculosis infection in US Holsteins. J Dairy Sci. 2006;89:1804–12.
Küpper J, Brandt H, Donat K, Erhardt G. Heritability estimates for Mycobacterium avium subspecies paratuberculosis status of German Holstein cows tested by fecal culture. J Dairy Sci. 2012;95:2734–9. https://doi.org/10.3168/jds.2011-4994.
van Hulzen KJE, Koets AP, Nielen M, Hoeboer J, van Arendonk JAM, Heuven HCM. Genetic variation for infection status as determined by a specific antibody response against Mycobacterium avium subspecies paratuberculosis in milk of Dutch dairy goats. J Dairy Sci. 2012;95:6145–51.
Kirkpatrick BW, Shook GE. Genetic susceptibility to Paratuberculosis. Vet Clin North Am Food Anim Pract. 2011;27:559–71.
Settles M, Zanella R, McKay SD, Schnabel RD, Taylor JF, Whitlock R, et al. A whole genome association analysis identifies loci associated with Mycobacterium avium subsp. paratuberculosis infection status in US holstein cattle. Anim Genet. 2009;40:655–62.
Pant SD, Schenkel FS, Verschoor CP, You Q, Kelton DF, Moore SS, et al. A principal component regression based genome wide analysis approach reveals the presence of a novel QTL on BTA7 for MAP resistance in Holstein cattle. Genomics. 2010;95:176–82.
Gonda MG, Kirkpatrick BW, Shook GE, Collins MT. Identification of a QTL on BTA20 affecting susceptibility to Mycobacterium avium ssp. paratuberculosis infection in US Holsteins. Anim Genet. 2007;38:389–96.
Zanella R, Settles ML, McKay SD, Schnabel R, Taylor J, Whitlock RH, et al. Identification of loci associated with tolerance to Johne’s disease in Holstein cattle. Anim Genet. 2011;42:28–38.
Minozzi G, Buggiotti L, Stella A, Strozzi F, Luini M, Williams JL. Genetic loci involved in antibody response to Mycobacterium avium ssp. paratuberculosis in cattle. PLoS ONE. 2010;5(6):e11117.
Kirkpatrick BW, Shi X, Shook GE, Collins MT. Whole-genome association analysis of susceptibility to paratuberculosis in Holstein cattle. Anim Genet. 2011;42:149–60.
Pant SD, Verschoor CP, Skelding AM, Schenkel FS, You Q, Biggar GA, et al. Bovine IFNGR2, IL12RB1, IL12RB2, and IL23R polymorphisms and MAP infection status. Mamm Genome. 2011;22:583–8.
Sharma BS, Abo-Ismail MK, Schenkel FS, You Q, Verschoor CP, Pant SD, et al. Association of TLR4 polymorphisms with Mycobacterium avium subspecies paratuberculosis infection status in Canadian Holsteins. Anim Genet. 2015;46:560–5.
Verschoor CP, Pant SD, You Q, Schenkel FS, Kelton DF, Karrow NA. Polymorphisms in the gene encoding bovine interleukin-10 receptor alpha are associated with infection status. BMC Genet. 2010;11:23.
Brito LF, Mallikarjunappa S, Sargolzaei M, Koeck A, Chesnais J, Schenkel FS, et al. The genetic architecture of milk ELISA scores as an indicator of Johne’s disease (paratuberculosis) in dairy cattle. J Dairy Sci. 2018;101:10062–75.
Lavers CJ, Barkema HW, Dohoo IR, McKenna SLB, Keefe GP. Evaluation of milk ELISA fordetection of Mycobacterium avium subspecies paratuberculosis indairy herds and association with within-herd prevalence. J Dairy Sci. 2014;97:299–309.
Grant IR. Zoonotic potential of Mycobacterium avium ssp . paratuberculosis : the current position. J Appl Microbiol. 2005;98:1282–93.
Tiwari A, Vanleeuwen JA, Mckenna SLB, Keefe GP, Barkema HW. Johne’s disease in Canada part I: clinical symptoms, pathophysiology, diagnosis, and prevalence in dairy herds. Can Vet J. 2006;47:874–82.
Mallikarjunappa S, Sargolzaei M, Brito LF, Meade KG, Karrow NA, Pant SD. Short communication: uncovering quantitative trait loci associated with resistance to Mycobacterium avium ssp. paratuberculosis infection in Holstein cattle using a high-density single nucleotide polymorphism panel. J Dairy Sci. 2018;101:7280–6.
Küpper J, Brandt H, Donat K, Erhardt G. Phenotype definition is a main point in genome-wide association studies for bovine Mycobacterium avium ssp. paratuberculosis infection status. Animal. 2014;8:1586–93.
Whittington RJ, Begg DJ, de Silva K, Purdie AC, Dhand NK, Plain KM. Case definition terminology for paratuberculosis (Johne’s disease). BMC Vet Res. 2017;13:1–13.
Buzanskas ME, Grossi DA, Ventura RV, Schenkel FS, Sargolzaei M, Meirelles SLC, et al. Genome-wide association for growth traits in canchim beef cattle. PLoS One. 2014;9(4):e94802.
Xing C, Xing G. Power of selective genotyping in genome-wide association studies of quantitative traits. BMC Proc. 2009;3:S23.
Li Y, Levran O, Kim JJ, Zhang T, Chen X, Suo C. Extreme sampling design in genetic association mapping of quantitative trait loci using balanced and unbalanced case-control samples. Sci Rep. 2009;9:15504.
Collins MT, Wells SJ, Petrini KR, Collins JE, Schultz RD, Whitlock RH. Evaluation of five antibody detection tests for diagnosis of bovine paratuberculosis. Clin and Diag Lab Immunol. 2005;12:685–92.
Kiser JN, White SN, Johnson KA, Hoff JL, Taylor JF, Neibergs HL. Identification of loci associated with susceptibility to mycobacterium avium subspecies paratuberculosis (map) tissue infection in cattle. J Anim Sci. 2017;95:1080–91.
Zare Y, Shook GE, Collins MT, Kirkpatrick BW. Genome-wide association analysis and genomic prediction of Mycobacterium avium subspecies paratuberculosis infection in US Jersey cattle. PLoS One. 2014;9:e88380.
Pauciullo A, Küpper J, Brandt H, Donat K, Iannuzzi L, Erhardt G. Wingless-type MMTV integration site family member 2 (WNT2) gene is associated with resistance to MAP in faecal culture and antibody response in Holstein cattle. Anim Genet. 2015;46:122–32.
Ruiz-Larrañaga O, Garrido JM, Iriondo M, Manzano C, Molina E, Montes I, et al. SP110 as a novel susceptibility gene for Mycobacterium avium subspecies paratuberculosis infection in cattle. J Dairy Sci. 2010;93:5950–8.
Pinedo PJ, Buergelt CD, Donovan GA, Melendez P, Morel L, Wu R, et al. Association between CARD15/NOD2 gene polymorphisms and paratuberculosis infection in cattle. Vet Microbiol. 2009;134:346–52.
Verschoor CP, Pant SD, You Q, Schenkel FS, Kelton DF, Karrow NA. Single nucleotide polymorphisms alter the promoter activity of bovine mif. Anim Biotechnol. 2011;22:143–50.
van Hulzen KJE, Schopen GCB, van Arendonk JAM, Nielen M, Koets AP, Schrooten C, et al. Genome-wide association study to identify chromosomal regions associated with antibody response to Mycobacterium avium subspecies paratuberculosis in milk of Dutch Holstein-Friesians. J Dairy Sci. 2012;95:2740–8.
Alpay F, Zare Y, Kamalludin MH, Huang X, Shi X, Shook GE, et al. Genome-wide association study of susceptibility to infection by Mycobacterium avium subspecies paratuberculosis in Holstein cattle. PLoS One. 2014;9.
Sallam AM, Zare Y, Alpay F, Shook GE, Collins MT, Alsheikh S, et al. An across-breed genome wide association analysis of susceptibility to paratuberculosis in dairy cattle. J D Res. 2017;84:61–7.
Feng Z, Wong WWL, Gao X, Schenkel F. Generalized genetic association study with samples of related individuals. Ann Appl Stats. 2011;5:2109–30.
Thornton TA. Statistical methods for genome-wide and sequencing association studies of complex traits in related samples. Curr Protoc Hum Genet. 2015;84:1.28.1–9.
VanRaden P. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91:4414–23.
Acknowledgements
We would sincerely like to thank Dr. Mehdi Sargolzaei (HiggsGene Solutions Inc.) for providing access to SNP1101 software. This work was presented at the International Society of Animal Genetics Conference in 2019 (https://www.isag.us/Docs/Proceedings/ISAG2019_Proceedings.pdf).
Funding
We would like to thank the following funding organizations for supporting this work: The Semex Alliance, Natural Sciences and Engineering Research Council (NSERC), and the Teagasc Walsh Fellowship for financial support to SM. The funding body did not have any role in in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
SM performed the analysis and wrote the first version of the manuscript; FS, LB and JC provided statistical guidance; LB estimated the sire EBVs; FM was involved in acquisition of data used for sire EBV estimation; NK, KM, NB, and ML designed the study, supervised it, and were involved in interpretation of the results. All authors read and revised the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No animal care committee approval was necessary for the purposes of this study, as no animal work was required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary File A:
List of SNPs used in the custom genotype panel.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mallikarjunappa, S., Schenkel, F.S., Brito, L.F. et al. Association of genetic polymorphisms related to Johne’s disease with estimated breeding values of Holstein sires for milk ELISA test scores. BMC Vet Res 16, 165 (2020). https://doi.org/10.1186/s12917-020-02381-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-020-02381-9