
Abstract
Background
Mucolipidosis II (ML II; I-cell disease) is caused by a deficiency of N-acetylglucosamine-1-phosphotransferase (GNPTAB; EC 2.7.8.17), which leads to a failure to internalize acid hydrolases into lysosomes for proper catabolism of various substances. This is an autosomal recessive lysosomal storage disease and causes severe progressive neuropathy and oculoskeletal dysfunction in humans (OMIM 252500). A naturally occurring disease model has been reported in juvenile domestic cats (OMIA 001248–9685) with clinical signs similar to human patients. We investigated the molecular genetic basis of ML II in a colony of affected cats by sequencing the coding and regulatory regions of GNPTAB from affected and clinically healthy related and unrelated domestic cats and compared the sequences to the published feline genome sequence (NCBI-RefSeq accession no. XM_003989173.4, Gene ID: 101100231).
Results
All affected cats were homozygous for a single base substitution (c.2644C > T) in exon 13 of GNPTAB. This variant results in a premature stop codon (p.Gln882*) which predicts severe truncation and complete dysfunction of the GNPTAB enzyme. About 140 GNPTAB variants have been described in human ML II patients, with 41.3% nonsense/missense mutations, nine occurring in the same gene region as in this feline model. Restriction fragment length polymorphism and allelic discrimination real-time polymerase chain reaction assays accurately differentiated between clear, asymptomatic carriers and homozygous affected cats.
Conclusion
Molecular genetic characterization advances this large animal model of ML II for use to further define the pathophysiology of the disease and evaluate novel therapeutic approaches for this fatal lysosomal storage disease in humans.
Similar content being viewed by others

Background
Mucolipidosis II (ML II, OMIM 252500) is a hereditary lysosomal storage disease caused by the deficiency of N-acetylglucosamine-1-Phosphotransferase (GNPTAB, EC 2.7.8.17, previously known as GNPTA). This enzyme is a heterohexamer composed of three homodimers: alpha (α), beta (β) and gamma (γ) in a molar ratio of 2:2:2. It is responsible for placing an integral targeting signal, a mannose 6-phosphate group, on newly synthesized lysosomal acid hydrolases inside the Golgi apparatus. This modification facilitates the transfer of the acid hydrolases into the lysosome compartments for the degradation of substances, such as oligosaccharides, lipids, and glycosaminoglycans (GAGs) [1, 2]. Thus, GNPTAB deficiency is associated with a unique deficiency of multiple enzymes in lysosomes rather than one specific enzyme deficiency, which then leads to the storage of various waste products, such as mucolipids and GAGs, within lysosomes. Those cells with the storage granules, known as “inclusion cells” or “I-cells”, can be identified microscopically and lent the disorder its original name “I-cell disease” in humans [3]. There is an infantile to juvenile age of onset with rapid progression of clinical signs in human patients, leading to death within the first decade of life. The clinical features of ML II include stunted growth, skeletal joint abnormalities, coarse facial features, corneal clouding, mental retardation, hepatomegaly, cardiomegaly and respiratory infections, similar to mucopolysaccharidoses (MPS). A clinically milder form of ML, ML III (OMIM 252600, also called ML III α/β, ML IIIA or Pseudo-Hurler polydystrophy), also exists. It is characterized by later onset (childhood) and slower progression [4, 5]. Although mucolipidosis I and IV were historically classified as mucolipidosis diseases, they are currently no longer considered part of the mucolipidosis family. Nonetheless, these storage disorders, along with ML II and III, are often grouped as ML (Table 1).
The three subunits (α, β and γ) of the GNPTAB enzyme complex are encoded by two genes. GNPTAB codes for the α and β subunits, and GNPTG codes for the γ subunit. In human patients, gene variants in GNPTAB cause ML II and ML III. Furthermore, GNPTG variants were discovered to also induce ML III clinically. Therefore, ML III was molecularly subtyped into an α/β form (ML IIIA) and a γ form (ML IIIC), where ML III α/β is due to GNPTAB pathogenic variants, while ML III γ is associated with variants in GNPTG [6, 7]. Currently, about 140 GNPTAB gene variants causing ML II and ML III α/β and 30 GNPTG variants causing ML III γ have been documented in human patients (The Human Gene Mutation Database, http://www.hgmd.cf.ac.uk).
In animals, ML II has been created in zebrafish [9, 10] and mice [11,12,13], and has been described as a naturally occurring disease in one domestic short-hair (DSH) cat from Switzerland (OMIA 001248–9685, ML II in Felis catus) [14, 15] which led to the clinicopathological characterization of the disease and the establishment of an animal colony at the School of Veterinary Medicine of the University of Pennsylvania [16]. No cases of ML III (either α/β or γ subtypes) have been documented to naturally occur in non-human species, but genetically engineered ML III γ disease models were created in zebrafish [17] and mice [13, 18].
Building upon initial work completed at the University of Pennsylvania which identified and characterized this naturally-occurring animal model of ML II, studies were undertaken to identify the molecular genetics of this disorder in the feline model. Those results are presented here.
Results
Clinical signs of ML II affected cats
One female (7160) and two males (7162 and 7163) out of five kittens born to clinically healthy DSH cats developed progressively deteriorating clinical signs shortly after birth. The three affected kittens showed very similar and typical clinical signs and disease courses, such as failure to thrive, coarse facial structures, short necks, ataxia, corneal clouding, behavioral dullness, and hepatomegaly. These severely progressive signs led to humane euthanasia within three months of life of affected kittens, while related carriers remained asymptomatic.
Biochemical enzymatic diagnosis assessment
The activities of six lysosomal enzymes were markedly increased in serum from the three affected kittens (α-L-iduronidase, 7-fold; arylsulfatase B, 19-fold; β-glucuronidase, 24-fold; α-D-mannosidase, 10-fold; α-D-fucosidase, 16-fold and N-acetyl-β-D-glucosaminidase, 7-fold) compared to ten age-matched, healthy, unrelated control kittens (Table 2). The assay values from affected kittens for all six enzymes were statistically significantly increased (p < 0.01) from affected kittens when compared to unrelated control cats. Similarly, assay values for all six enzymes were also statistically significantly increased (p < 0.01) from affected kittens when compared to a combined group of unrelated control cats and clinically healthy littermates (n = total of 12). No statistically significant difference was found when comparing clinically healthy littermates to unrelated controls.
Note, these enzyme activities were not measured in lysosomes (previously shown to be deficient [16]), and GNPTAB enzyme activity was not measured due to a lack of available substrate.
GNPTAB gene sequence and nonsense variant identification
The feline GNPTAB gene is on chromosome B4 and consists of 21 exons with a coding sequence length of 1256 amino acid residues for the α and β subunits of the GNPTAB protein. The amino acid sequence homology of GNPTAB between felines (NCBI-ReqSeq accession no. XP_003989222.1, NCBI Genbank) and humans (NCBI-ReqSeq accession no. NP_077288, NCBI Genbank) is 91%.
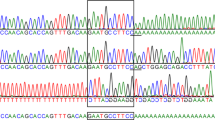
Comparing the GNPTAB sequences of the three ML II affected kittens to an unrelated clinically healthy cat (9802) and published feline genome sequences, revealed one homozygous single nucleotide substitution in exon 13 c.2644C > T (Fig. 1). This base change results in the conversion of CAG, which is coding for glutamine (Gln), to TAG, a premature stop codon, at protein position 882 (p.Gln882*) causing truncation of the GNPTAB α/β-subunits. Furthermore, the clinically healthy parents of the affected kittens (tom 4977 and queen 6431) were heterozygous for the GNPTAB variant and wild type alleles while the clinically healthy littermates (7161 and 7164), which had normal serum lysosomal enzyme activities, and one clinically healthy unrelated cat (9802) were homozygous for the wild type allele. No other gene variants were found in the coding sequence of GNPTAB in affected kittens and their family members when compared to the published feline genome sequence of GNPTAB.

The feline GNPTAB structure and DNA sequencing results surrounding the c.2644C > T variation in exon 13 which is predicted to produce a GNPTAB protein without a β subunit. a A phenotypically healthy littermate (7161), showing homozygosity for cytosine (c.2644C, arrow). b A ML II affected kitten (7160), revealing homozygosity for a C > T substitution (c.2644 T), which will change the codon CAG (for glutamine) to TAG (a stop codon) at GNPTAB protein position 882 (p.Gln882*). c The clinically healthy tom cat (4977) of the ML II affected kittens, displaying both wild type and mutant allele (c.2644Y, heterozygous status). d Feline GNPTAB protein domain structure (adopted from human construct with 91% amino acids homology between human and cats). e The ML II affected cats with the p.Gln882* variant predicted to produce a GNPTAB enzyme with a truncated α subunit and without β subunit
Genotyping assays
In a restriction fragment length polymorphism (RFLP) genotyping assay, ML II affected kittens showed only the 414 base pairs (bp) uncut fragment, while their parents had three DNA fragments (carrier pattern; 414, 231 and 183 bp), and the two clinically healthy littermates and the clinically healthy unrelated cat (9802) revealed two fragments (183 and 231 bp) (Fig. 2b). Similarly, the mutant variant segregated completely in the family pedigree (Fig. 2a), confirming an autosomal recessive inheritance pattern.

Pedigree and PCR-RFLP allele specific analyses within a family of the ML II affected cats. a Pedigree of a feline family descended from the first reported ML II affected cat [14, 15]. #3739 was the clinically asymptomatic half-brother of the originally affected cat. The family pedigree illustrates complete concordance and an autosomal recessive pattern. b Results of PCR-RFLP c.2644C > T allele specific analyses. A DNA fragment surrounding the variant was amplified by PCR, digested with PstI restriction enzyme, and visualized on 2% agarose gel stained with ethidium bromide. The amplified product is 414 bp in length, which includes one PstI digestion site. The clinically healthy kittens (7161 and 7164, and the normal control 9802) produced two fragments of 231 and 183 bp. The three ML II affected kittens (7160, 7162 & 7163) only showed an un-digested 414 bp fragment. The clinically healthy tom (4977) and queen (6431) were heterozygous for the wild type and variant alleles and therefore exhibited all three fragments of 414, 231 and 183 bp
The allelic discrimination real-time polymerase chain reaction (PCR) assay for the c.2644C > T nonsense variant in the GNPTAB gene clearly differentiated the three genotypes (Additional file 3 Figure S1): the ML II affected cats 7160, 7162 and 7163 were homozygous for the variant allele, presenting in the top left corner of the allelic discrimination plot; two clinically non-affected littermates (7161 and 7164) and one clinically healthy unrelated cat (9802) were homozygous for the wild type alleles, shown in the lower right corner of the plot; clinically non-symptomatic tom (4977) and queen (6431) exhibited the heterozygote genotype which appear in the center of the plot. Furthermore, there was complete segregation of the mutant GNPTAB allele among five ML II affected, eight obligate carriers and 50 healthy cats within the colony. Finally, 100 unrelated DSH cats from the local Pennsylvania region did not have the mutant GNPTAB allele, and no other naturally occurring cases of ML II have been reported since the original kitten.
Discussion
This enzymatic and molecular genetic study of a related group of DSH cats clinically affected with ML II identifies the genetic defect causative of ML II to be a single nonsense variant in GNPTAB. Cats are the only naturally occurring large animal model of ML II identified to date, and have clinical features very similar to ML II in human patients, including failure to thrive, behavioral dullness, facial dysmorphia, retinal changes, decreased muscle tone, and ataxia, all of which are present at a young juvenile age (within two months of birth) with rapid progression. Thus, the molecular genetics, biochemical, and clinical abnormalities reported in these cats appear homologous with those of ML II in human patients [4, 5, 20], making this lineage of cats an ideal disease model to investigate therapeutic interventions.
Clinically, ML II presents similarly to other lysosomal storage diseases such as MPS, and the diagnosis must be confirmed by biochemical enzymatic and/or molecular genetic determinations [4]. Ideally, the biochemical diagnosis for ML II disease would directly detect the lack of GNPTAB activity in the white blood cells or cultured fibroblasts, but this enzymatic assay requires a radioactive substrate which is not commercially available. Instead, the diagnosis is typically reached by measuring the high activities of all lysosomal acid hydrolases in serum [16, 21]. This is because the pathogenic mechanism of ML II results in a defect in cellular trafficking of acid hydrolases, preventing intracellular transport of these enzymes into lysosomes, and resulting in the secretion of the enzymes extracellularly into the serum. In the study reported here, the enzymes from three cats with ML II were 7 to 24-fold above normal serum activities, values similar to what has been observed in human patients with ML II [21]. In previous work, we have demonstrated the lack of acid hydrolase enzymes in lysosomes of kittens with ML II [16].
Any gene variants affecting GNPTAB expression, secretion, and function could result in a lack of enzymatic GNPTAB activity and cause ML II or III α/β disorders. In this study of a family of cat with ML II, a single novel homozygous nonsense variant (c.2644C > T) in exon 13 was discovered in the GNPTAB coding sequence. This variant creates a premature stop codon (p.Gln882*).
The cleavage site of the α and β subunit precursor is located in exon 14 between residue 928, a lysine, and 929, an aspartic acid [22], in both humans and cats (Fig. 1d). It is predicted that the feline c.2644C > T, p.Gln882* nonsense variant would cause premature decay of the truncated messenger RNA of the GNPTAB α subunit, and a complete lack of the β subunit (Fig. 1e). The expected phenotype and pathogenic mechanism of action from a defective GNPTAB protein resulting from this truncated RNA message is consistent with the clinical signs and biochemical parameters observed in these cats. The clinical presentation and disease course due to this nonsense variant was completely penetrant in all affected ML II kittens in this study and very similar to previous reports [14,15,16].
While this p.Gln882* nonsense variant has not been described in human patients with ML II, nine nonsense/missense variants in this gene region (exon 13) have been documented in human patients with a severe ML II phenotype (Table 3), indicative of similar phenotype with mis- or nonsense variants in the same gene region. All nine variations have caused ML II with early progressive clinical symptoms in human patients [20, 23,24,25,26,27], similar to the feline disease model described here.
Genotyping of ML II affected cats and their family members and comparison to unrelated cats confirmed that feline ML II is inherited as an autosomal recessive trait (Fig. 2a). Moreover, it verified the lack of the ML II genetic variant within the greater population of cats. Obligate and other carriers appeared clinically healthy but were readily detected by genotyping tests and were all related to the common ancestor. While this specific variation is not expected to be widespread in the feline population, the established RFLP and real-time PCR assays allow precise genotyping of animals in this research colony for assessment of novel therapeutic interventions.
This study builds upon a previously published abstract within which we reported preliminary molecular studies on GNPTAB in ML II cats [28]. At the time of the abstract’s publication, both the affected cat’s GNPTAB sequence and the feline genome reference sequence were incomplete. Complete sequencing of the affected cat’s GNPTAB exome, as reported here, as well as publication of the complete feline genome reference sequence, revealed that the GNPTAB missense variant referred to in the research abstract is, in fact, correctly a nonsense variant.
Conclusion
Identification of the molecular basis of ML II in cats provides further evidence of its similarity to the human disease and enhances the use of this unique disease model to further define the pathogenesis of ML II and to evaluate safety and efficacy of novel therapeutic approaches for this lysosomal disorder in children.
Methods
Animals and phenotyping
Cats genetically-related to the originally-described cat with naturally-occurring mucolipidosis II identified in Switzerland [14, 15] have been a part of the animal colony at the School of Veterinary Medicine University of Pennsylvania for the past decade, and are cared for according to guidelines for animal research. The clinically non-affected half-brother (3739) of the originally affected cat was their common ancestor (Fig. 2a). A family (tom, queen, and five offspring including three ML II clinically affected kittens) was selected for this investigation. The clinically non-affected tom (4977) and queen (6431) produced five kittens with two clinically healthy (7161 and 7164 both male) and three ML II affected (7160 female, 7162 and 7163 both male) kittens. Unrelated age-matched (2–4 months) DSH cats kept in the same animal colony served as controls. Mucolipidosis II affected kittens were humanely euthanized at three months of age due to the severity of clinical manifestations. Euthanasia was performed with Euthasol euthanasia solution (Virbac AH, Inc., Fort Worth, TX, USA) at the dosage of 80 mg/kg intravenously according to the American Veterinary Medical Association Guidelines for the Euthanasia of Animals. The clinically asymptomatic cats remained in the colony or were adopted. The research methods and euthanasia protocol were approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania (IACUC number A3078–01). Serum and ethylenediaminetetraacetic acid (EDTA) anticoagulated blood samples were collected from each cat for this study. Other DNA samples were obtained from a DNA bank at the University of Pennsylvania.
Biochemical enzymatic diagnosis
The enzymatic activities of six lysosomal enzymes were measured in serum of affected and related kittens and compared to the enzymatic activities of ten age-matched clinically healthy unrelated cats. The tested enzymes assays were performed in duplicates for each sample according to the standard protocols utilizing the 4-Methylumbelliferone (4-MU) fluorometric endpoint method [29, 30] and as previously described by the PennGen laboratory [16] (Additional file 1). Enzyme activity values were expressed as nMol of 4-MU produced per mL of serum per hour.
Statistical analysis of the biochemical assay results from cats with ML II, unrelated control cats, and clinically healthy littermate, was performed. To overcome the non-heterogeneous range, the results were compared utilizing median values but not average values, using the Mann-Whitney U-test (two samples nonparametric tests, GenStat, version 18; VSN international ltd) [31]. P-values of less than 0.05 were considered significant. Results of the assays from cats with ML II were compared to those of the unrelated control cats for all six enzymes. Further analysis compared results from affected cats with a combined group of unrelated control cats and clinically healthy littermates, as well as comparison of unrelated control cats to clinically healthy littermates.
Genomic DNA sequencing and variant identification
Genomic DNA (gDNA) was extracted from EDTA-anticoagulated blood from the tom and queen, and their five offspring, and a clinically healthy unrelated cat (9802) using QIAamp DNA Blood Mini kit (QIAGEN, Valencia, CA) following the manufacturer’s standard protocol.
The feline GNPTAB gene with its 21 exons as well as exon-intron boundaries, and its 5′- and 3′- untranslated regions, were amplified using 20 primer pairs and hot-start PCR (KOD Xtreme™ Hot Start DNA Polymerase kit, Novagen-EMD company, Gibbstown, NJ) following the laboratory’s established protocol [32]. The PCR primers were designed according to the published feline genome sequences (NCBI-RefSeq accession no. XM_003989173.4, Gene ID: 101100231, NCBI Genbank https://www.ncbi.nlm.nih.gov and Gene ID: ENSFCAG00000008281; Transcript ID: ENSFCAT00000008283.3, Felis_catus_6.2 reference genome assembly, Ensembl http://www.ensembl.org) and synthesized by Integrated DNA Technologies, Inc. (IDT, Coralville, IA) (Additional file 2). A negative control containing DNA-free water instead of gDNA was included in all assays to detect any possible contamination.
The PCR products were purified [32] and Sanger sequenced at the DNA Sequencing Core Facility of Perelman School of Medicine at the University of Pennsylvania: 6 μL (10 ng/μL) of template DNA for both forward and reverse primer tubes with 3 μL (1.1 μM) of each primer. The sequencing results were analyzed and compared to the published feline genome sequences as well as to the sequencing results of the clinically healthy cat (Lasergene software, DNAStar, Inc., Madison, WI).
Genotyping assays
Based upon the discovery of a single nonsense variant in the GNPTAB gene in ML II affected cats, genotyping tests were established. For the RFLP assay, PCR primers surrounding the variant allele region (exon 13) were used (Additional file 2) which produced an amplicon of 414 bp followed by a PstI endonuclease (5′...CTGCA^G…3′, New England Biolabs, Ipswich, MA) digestion. Ten μL of the digested reaction mixture, which included PstI (10,000 U/mL), NEBuffer 3.1 (10X), and DNA-free water at a ratio of 0.5:2.0:7.5, was mixed with 10 μL of the PCR product in a 37 °C water bath overnight. The PCR product of the normal allele was digested into two fragments of 231 and 183 bp, which were visualized on a 2% agarose gel stained with ethidium bromide. The mutant variant allele removed the PstI recognition site and thereby prevented the digestion, resulting in a single 414 bp fragment.
For the allelic discrimination real-time PCR assay, reagents were obtained from the Custom Taqman SNP Genotyping Assay (Applied Biosystems, Life Technologies, Grand Island, NY). The DNA fragment around the nonsense variant was amplified by real-time PCR using a 15 μL reaction mixture containing 7.5 μL of 2X TaqMan Universal PCR master mix, 0.375 μL of 40X assay mix consisting of unlabeled PCR primers (Additional file 2), and fluorescently labeled TaqMan 3′-Minor groove binder probes (VIC dye-labeled CGTGATGCTGCAGTTT for wild type, and FAM dye-labeled CGTGATGCTACAGTTT for c.2644C > T substitution allele), 6.125 μL of DNA-free water, and 1 μL of template gDNA. Standard thermal cycling parameters were used: 95 °C for 10 min for an initial heat, 40 cycles of 15 s at 95 °C for denaturation and 60 s at 60 °C for annealing/extension, and 1 min at 60 °C for post-PCR hold. Results of amplification and detection were expressed as end-point fluorescence intensities (Rn values) plotted between wild type allele (X-axis) and variant allele (Y-axis) quadrants (Applied Biosystems ABI 7500 instrument, Life Technologies, Grand Island, NY).
The three ML II affected cats and their family members reported in this study (parents, obligate carriers [n = 2]; clinically asymptomatic littermates [n = 2]), and a clinically healthy unrelated cat were tested with both genotyping assays. An additional five ML II affected cats, eight obligate carriers, 50 healthy cats within the research colony and 100 unrelated DSH cats from the local Pennsylvania region were only tested by allelic discrimination real-time PCR assay.
Abbreviations
- 4-MU:
-
4-Methylumbelliferone
- Bp:
-
base pairs
- DSH:
-
domestic short-hair cat
- EDTA:
-
ethylenediaminetetraacetic acid
- GAGs:
-
glycosaminoglycans
- gDNA:
-
genomic DNA; Gln, glutamine
- GNPTAB:
-
N-acetylglucosamine-1-phosphotransferase
- HMD:
-
The Human Gene Mutation Database
- IQR:
-
interquartile range
- ML II:
-
mucolipidosis II
- MPS:
-
mucopolysaccharidoses
- OMIA:
-
Online Mendelian Inheritance in Animal
- OMIM:
-
Online Mendelian Inheritance in Man
- OMMBID:
-
Online Metabolic and Molecular Bases of Inherited Disease
- PCR:
-
polymerase chain reaction
- RFLP:
-
restriction fragment length polymorphism
References
Qian Y, Lee I, Lee WS, Qian M, Kudo M, Canfield WM, et al. Functions of the α, β, and γ subunits of UDP-GlcNAc:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. J Biol Chem. 2010;285:3360–70. https://doi.org/10.1074/jbc.M109.068650.
Natowicz MR, Chi MM, Lowry OH, Sly WS. Enzymatic identification of mannose 6-phosphate on the recognition marker for receptor-mediated pinocytosis of 8-glucuronidase by human fibroblasts. Proc Natl Acad Sci U S A. 1979;76(9):4322–6. 291966. https://doi.org/10.1073/pnas.76.9.4322.
Tondeur M, Vamos-Hurwitz E, Mockel-Pohl S, Dereume JP, Cremer N, Loeb H. Clinical, biochemical, and ultrastructural studies in a case of chondrodystrophy presenting the I-cell phenotype in tissue culture. J Pediatr. 1971;79(3):366–78. 4327937. https://doi.org/10.1016/S0022-3476(71)80143-9.
Braulke T, Raas-Rothschild A, Kornfeld S. I-cell disease and pseudo-hurler Polydystrophy: disorders of lysosomal enzyme phosphorylation and localization. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, et al., editors. The online metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2013. Chapter 138. Available: http://ommbid.mhmedical.com/book.aspx?bookid=971.
Cathey SS, Kudo M, Tiede S, Raas-Rothschild A, Braulke T, Beck M, et al. Molecular order in mucolipidosis II and III nomenclature. Am J Med Genet. 2008;146A(4):512–3. https://doi.org/10.1002/ajmg.a.32193.
Encarnacao M, Lacerda L, Costa R, Prata MJ, Coutinho MF, Ribeiro H, et al. Molecular analysis of the GNPTAB and GNPTG genes in 13 patients with mucolipidosis type II or type III - identification of eight novel mutations. Clin Genet. 2009;76:76–84. https://doi.org/10.1111/j.1399-0004.2009.01185.x 19659762.
Braulke T, Pohl S, Storch S. Molecular analysis of the GlcNac-1-phosphotransferase. J Inherit Metab Dis. 2008;31:253–7. https://doi.org/10.1007/s10545-008-0862-5.
Rottier RJ, Bonten E. d'Azzo a. a point mutation in the neu-1 locus causes the neuraminidase defect in the SM/J mouse. Hum Mol Genet. 1998;7(2):313–21. https://doi.org/10.1093/hmg/7.2.313 9425240.
Flanagan-Steet H, Sias C, Steet R. Altered chondrocyte differentiation and extracellular matrix homeostasis in a zebrafish model for mucolipidosis II. Am J Pathol 2009 Nov;175(5):2063–2075. https://doi.org/10.2353/ajpath.2009.090210 PMID: 19834066.
Petrey AC, Flanagan-Steet H, Johnson S, Fan X, De la Rosa M, Haskins ME, et al. Excessive activity of cathepsin K is associated with cartilage defects in a zebrafish model of mucolipidosis II. Dis Model Mech. 2012;5:177–90. 22046029. https://doi.org/10.1242/dmm.008219.
Kollmann K, Damme M, Markmann S, Morelle W, Schweizer M, Hermans-Borgmeyer I, et al. Lysosomal dysfunction causes neurodegeneration in mucolipidosis II “knock-in” mice. Brain 2012:135(9):2661–2675. https://doi.org/10.1093/brain/aws209 PMID: 22961545.
Paton L, Bitoun E, Kenyon J, Priestman DA, Oliver PL, Edwards B, et al. A novel mouse model of a patient mucolipidosis II mutation recapitulates disease pathology. J Biol Chem. 2014;289(39):26709–21. 25107912. https://doi.org/10.1074/jbc.M114.586156.
Idol RA, Wozniak DF, Fujiwara H, Yuede CM, Ory DS, Kornfeld S, et al. Neurologic abnormalities in mouse models of the lysosomal storage disorders mucolipidosis II and mucolipidosis III γ. PLoS One. 2014;9(10):e109768. 25314316. https://doi.org/10.1371/journal.pone.0109768.
Bosshard NU, Hubler M, Arnold S, Briner J, Spycher MA, Sommerlade HJ, et al. Spontaneous mucolipidosis in a cat: an animal model of human I-cell disease. Vet Pathol. 1996;33(1):1–13. https://doi.org/10.1177/030098589603300101 8826001.
Hubler M, Haskins ME, Arnold S, Kaser-Hotz B, Bosshard NU, Briner J, et al. Mucolipidosis type II in a domestic shorthair cat. J Small Anim Pract 1996;37(9):435–441. PMID: 8887204.
Mazrier H, Van Hoeven M, Wang P, Knox VM, Aguirre GD, Holt E, et al. Inheritance, biochemical abnormalities, and clinical features of feline mucolipidosis II: the first animal model of human I-cell disease. J Hered. 2003;94(5):363–73. 14557388. https://doi.org/10.1093/jhered/esg080.
Flanagan-Steet H, Matheny C, Petrey A, Parker J, Steet R. Enzyme-specific differences in mannose phosphorylation between GlcNAc-1-phosphotransferase αβ and γ subunit deficient zebrafish support Cathepsin proteases as early mediators of mucolipidosis pathology. Biochim Biophys Acta. 2016;1860(9):1845–53. 27241848. https://doi.org/10.1016/j.bbagen.2016.05.029.
Vogel P, Payne BJ, Read R, Lee W-S, Gelfman CM, Kornfeld S. Comparative pathology of murine mucolipidosis types II and IIIC. Vet Pathol. 2009;46(2):313–24. https://doi.org/10.1354/vp.46-2-313 PMID: 19261645.
Fares H, Greenwald I. Regulation of endocytosis by CUP-5, the Caenorhabditis elegans mucolipin-1 homolog. Nat Genet 2001;28(1):64–68. PMID: 11326278.
Cathey SS, Leroy JG, Wood T, Eaves K, Simensen RJ, Kudo M, et al. Phenotype and genotype in mucolipidoses II and III alpha/beta: a study of 61 probands. J Med Genet. 2010;47:38–48. 19617216. https://doi.org/10.1136/jmg.2009.067736.
Sprigz RA, Doughty RA, Spackman TJ, Murnane MJ, Coates PM, Koldovsky O, et al. Neonatal presentation of I-cell disease. J Pediatr. 1978;93:954–8. 722439. https://doi.org/10.1016/S0022-3476(78)81218-9.
Coutinho MF. N-acetylglucosamine-1-phosphate transferase, alpha/beta and gamma subunits (GNPTAB, GNPTG). In: Taniguchi N, Honke K, Fukuda M, Narimatsu H, Yamaguchi Y, Angata T, editors. Handbook of glycosyltransferases and related genes. Tokyo: Springer; 2014. p. 1335–47. https://doi.org/10.1007/978-4-431-54240-7.
Fernández-Marmiesse A, Morey M, Pineda M, Eiris J, Couce ML, Castro-Gago M, et al. Assessment of a targeted resequencing assay as a support tool in the diagnosis of lysosomal storage disorders. Orphanet J Rare Dis. 2014;9:59. 24767253. https://doi.org/10.1186/1750-1172-9-59.
Kang C, Riazuddin S, Mundorff J, Krasnewich D, Friedman P, Mullikin JC, et al. Mutations in the lysosomal enzyme–targeting pathway and persistent stuttering. N Engl J Med 2010;362:677–685. https://doi.org/10.1056/NEJMoa0902630 PMID: 20147709.
Zarghooni M, Dittakavi SS. Molecular analysis of cell lines from patients with mucolipidosis II and mucolipidosis III. Am J Med Genet. 2009;149A:2753–61. 19938078. https://doi.org/10.1002/ajmg.a.33134.
Tiede S, Storch S, Lübke T, Henrissat B, Bargal R, Raas-Rothschild A, et al. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat Med 2005;11(10):1109–1112. https://doi.org/10.1038/nm1305 PMID: 16200072.
Paik KH, Song SM, Ki CS, Yu HW, Kim JS, Min KH, et al. Identification of mutations in the GNPTA (MGC4170) gene coding for GlcNAc-phosphotransferase alpha/beta subunits in Korean patients with mucolipidosis type II or type IIIA. Hum Mutat. 2005;26:308–14. 16116615. https://doi.org/10.1002/humu.20205.
Giger U, Tcherneva E, Caverly J, Seng A, Huff AM, Cullen K, et al. A missense point mutation in N-acetylglucosamine-1-phospotrans-ferase causes mucolipidosis II in domestic shorthair cats. J Vet Intern Med. 2006;20:781.
Wenger DA, Williams C. Screening for lysosomal disorders. In: Hommes FA, editor. Techniques in diagnostic human biochemical genetics. A laboratory manual. New York: Wiley-Liss; 1991. p. 587–617.
Chang PL, Rosa NE, Davidson RG. Differential assay of arylsulfatase a and B activities: a sensitive method for cultured human cells. Anal Biochem. 1981;117(2):382–9. 6119929. https://doi.org/10.1016/0003-2697(81)90795-8.
GenStat VSN International, https://www.vsni.co.uk/software/genstat/. Accessed December 2017.
Wang P, Sorenson J, Strickland S, Mingus C, Haskins ME, Giger U. Mucopolysaccharidosis VII in a cat caused by 2 adjacent missense mutations in the GUSB gene. J Vet Intern Med. 2015;29:1022–8. 26118695. https://doi.org/10.1111/jvim.13569.
Acknowledgements
The authors thank Dr. Eva Tcherneva for her initial laboratory studies on this project and appreciate the work of the animal colony staff.
Funding
This study was supported by the National Institutes of Health, grant number OD 010939. This grant sponsors the research of animal models of human genetic disease and funds the animal colony, required materials and the staff. The sponsors had no involvements in the design of the study, analysis and interpretation of data, and in writing the manuscript.
Availability of data and materials
All data generated and analyzed during this study are included in this published article and its supplementary information files.
Author information
Authors and Affiliations
Contributions
Research concept and design: PW, HM, JCR and UG. Statistical analysis: HM. Samples collection and experiment performance: PW. Data analysis, verification and interpretation: PW, KJ and UG. Clinical investigation: HM and JCR. Research management and coordination: UG. Original manuscript preparation: PW and UG. Manuscript writing: all authors. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Authors’ information
Dr. Hamutal Mazrier’s current address is The University of Sydney, Faculty of Science, Sydney School of Veterinary Science, NSW 2006, Australia. Dr. Jessica Caverly Rae’s current address is GlaxoSmithKline, Collegeville, PA 19426, USA.
Ethics approval and consent to participate
The research was approved by the Institutional Animal Care and Use Committee at the University of Pennsylvania (IACUC number A3078–01).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Lysosomal enzymes assays: substrates, buffers, conditions and references. (DOCX 15 kb)
Additional file 2:
Table S2. PCR primers and conditions used for DNA sequencing of the feline GNPTAB exons. (DOCX 25 kb)
Additional file 3:
Figure S1. Allelic discrimination assay detects c.2644C > T variant in a cat family with ML II kittens. (TIF 2756 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Wang, P., Mazrier, H., Caverly Rae, J. et al. A GNPTAB nonsense variant is associated with feline mucolipidosis II (I-cell disease). BMC Vet Res 14, 416 (2018). https://doi.org/10.1186/s12917-018-1728-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12917-018-1728-1