
Abstract
Background
The effect of the combination of an anti-angiogenic agent with a poly (ADP-ribose) polymerase (PARP) inhibitor in cancer treatment is unclear. We assessed the oral combination of fuzuloparib, a PARP inhibitor, and apatinib, a VEGFR2 inhibitor for treating advanced ovarian cancer (OC) or triple-negative breast cancer (TNBC).
Methods
This dose-escalation and pharmacokinetics-expansion phase 1 trial was conducted in China. We used a standard 3 + 3 dose-escalation design, with 7 dose levels tested. Patients received fuzuloparib orally twice daily, and apatinib orally once daily. The study objectives were to determine the safety profile, recommended phase 2 dose (RP2D), pharmacokinetics, preliminary efficacy, and efficacy in relation to germline BRCA mutation (gBRCAmut).
Results
Fifty-two pre-treated patients were enrolled (30 OC/22 TNBC). 5 (9.6%) patients had complete response, 14 (26.9%) had partial response, and 15 (28.8%) had stable disease. Objective response rate (ORR) and disease control rate were 36.5% (95% CI 23.6–51.0) and 65.4% (95% CI 50.9–78.0), respectively. At the highest dose level of fuzuloparib 100 mg plus apatinib 500 mg, the ORR was 50.0% (4/8; 95% CI 15.7–84.3); this dose was determined to be the RP2D. Patients with gBRCAmut had higher ORR and longer median progression-free survival (PFS) than those with gBRCAwt, both in OC (ORR, 62.5% [5/8] vs 40.9% [9/22]; PFS, 9.4 vs 6.7 months) and TNBC (ORR, 66.7% [2/3] vs 15.8% [3/19]; PFS, 5.6 vs 2.8 months). Two dose-limiting toxicities occurred: grade 4 febrile neutropenia (fuzuloparib 100 mg plus apatinib 250 mg) and thrombocytopenia (fuzuloparib 100 mg plus apatinib 375 mg). Maximum tolerated dose was not reached. The most common treatment-related grade ≥ 3 toxicities in all patients were hypertension (19.2%), anaemia (13.5%), and decreased platelet count (5.8%). Exposure of apatinib increased proportionally with increasing dose ranging from 250 to 500 mg, when combined with fuzuloparib 100 mg.
Conclusions
Fuzuloparib plus apatinib had acceptable safety in patients with advanced OC or TNBC. Fuzuloparib 100 mg bid plus apatinib 500 mg qd was established as the RP2D. With the promising clinical activity observed, this combination is warranted to be further explored as a potential alternative to chemotherapy.
Trial registration
ClinicalTrials.gov, NCT03075462 (Mar. 9, 2017).
Similar content being viewed by others
Background
According to the Cancer Genome Atlas, aggressive tumors such as high-grade serous ovarian cancer (HGSOC) and triple-negative breast cancer (TNBC) exhibit genomic characteristics associated with deficiencies in the homologous recombination (HR) repair pathway (e.g. BRCA deficiency) [1, 2]. Poly (ADP-ribose) polymerase (PARP) inhibitors cause the accumulation of double-strand breaks that cannot be effectively repaired, leading to synthetic lethality in HR-deficient cells [3, 4]. PARP inhibitors are currently indicated as treatment for BRCA-mutated (BRCAmut) OC and HER2- BC, as well as maintenance treatment for OC after response to platinum-based chemotherapy, regardless of BRCAmut [5,6,7]. Therefore, BRCA detection becomes particularly important. To further expand the scope of potential patients who could benefit from PARP inhibitors (e.g. those with platinum-resistant, non-BRCAmut disease) and to spare the toxicities from repeated platinum therapy, there is a need for exploration of effective chemo-free combination regimens.
Anti-angiogenic therapy could induce hypoxia in the tumor microenvironment, cause genetic instability and downregulation of BRCA1/2 and lead to HR deficiency, thereby potentiating response to PARP inhibitors [8,9,10]. The combination of anti-angiogenic therapy and a PARP inhibitor has been evaluated in several clinical trials. In phase 2 trials of platinum-sensitive recurrent OC, the addition of the anti-vascular endothelial growth factor (VEGF) antibody bevacizumab to niraparib, or the VEGF receptor (VEGFR) inhibitor cediranib to olaparib, improved progression-free survival (PFS) [11,12,13]. Promising anti-tumor activity was also observed with the olaparib-cediranib combination in patients with platinum-resistant OC, regardless of BRCA mutation status [14,15,16]. However, the administration of bevacizumab requires intravenous infusion, which may decrease the convenience of treatment. In addition, although cediranib is an oral tyrosine kinase inhibitor (TKI), the olaparib-cediranib combination has been associated with tolerability issues, leading to dose reduction in over 70% of OC patients [13].
Fuzuloparib (formerly known as fluzoparib) is a novel PARP inhibitor. It has been approved in China as treatment for germline BRCA1/2-mutated (gBRCA1/2mut), platinum-sensitive recurrent OC, and as maintenance therapy for platinum-sensitive recurrent OC, regardless of BRCA1/2 mutation status [17,18,19]. In phase 2 and 3 trials, fuzuloparib was well tolerated in OC patients, with a low treatment discontinuation rate due to adverse events (AEs) [17, 18]. Compared with other approved PARP inhibitors, fuzuloparib has a lower risk of gastrointestinal toxicities, which might be related to its postprandial administration and high oral bioavailability [20]. Apatinib is a highly selective VEGFR2-targeted TKI. It has demonstrated efficacy and safety across a wide variety of solid tumors, including OC and BC [21,22,23,24]. In a murine xenograft model, the combination of fuzuloparib and apatinib showed enhanced antitumor efficacy compared with apatinib alone, without apparent incremental toxicity [20]. Currently, the oral combination of fuzuloparib and apatinib is undergoing clinical development, aiming to improve treatment convenience and spare patients from the toxicities associated with conventional chemotherapy. In this phase 1 trial, we assessed the safety, clinical activity, and pharmacokinetics (PK) of fuzuloparib plus apatinib in the treatment of patients with advanced OC or TNBC. Furthermore, we conducted biomarker analysis according to gBRCA1/2 mutation.
Methods
This multi-center, open-label, dose-escalation and PK-expansion phase 1 study (ClinicalTrials.gov, number NCT03075462) was conducted in 10 centers in China. Patients enrolled were women aged 18–70 years, had confirmed advanced high-grade serous epithelial ovarian, fallopian tube, or primary peritoneal cancer (with or without gBRCAmut), or TNBC. Patients with OC were required to have received 2–4 prior lines of platinum-based chemotherapy. Both platinum-sensitive (disease progression or relapse ≥ 6 months after the last platinum-based chemotherapy) or resistant (disease progression or relapse within 1–6 months after the last platinum-based therapy) disease were allowed. Patients with TNBC were required to receive ≤ 2 prior lines of chemotherapy for advanced disease, and had disease progression or recurrence during or after the last anti-cancer treatment. Other inclusion criteria were an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1, a life expectancy of ≥ 3 months, measurable disease per Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 and adequate organ functions. Key exclusion criteria were prior use of PARP inhibitors or anti-angiogenic agents, central nervous system metastases, uncontrolled hypertension (systolic blood pressure > 150 mmHg or diastolic blood pressure > 90 mmHg), a history of congestive heart failure, bowel obstruction or gastrointestinal bleeding (grade 3 or 4 per Common Terminology Criteria for Adverse Events [CTCAE] v4.03) within 4 weeks.
The trial protocol was approved by the institutional review board or ethnic review committee of each participating center. All procedures were conducted in accordance with the guidelines of Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent before enrollment.
Procedure
The study used a standard 3 + 3 dose escalation design. The starting dose of fuzuloparib was 40 mg twice daily (bid) in combination with a fixed dose of apatinib at 250 mg once daily (qd); fuzuloparib could be dose escalated in 20 mg increments to up to 100 mg. An additional dose level of fuzuloparib 80 mg bid plus apatinib 375 mg qd was planned after assessment of the regimen of fuzuloparib 80 mg bid plus apatinib 250 mg qd. If dose escalation of fuzuloparib to 100 mg was completed, dose escalation for apatinib was initiated with two dose level planned: 375 mg qd and 500 mg qd (with fuzuloparib fixed at 100 mg bid). During dose escalation, the next-higher dose level was opened if less than one-third of patients in the previous dose level experienced a dose-limiting toxicity (DLT). Based on preliminary safety and efficacy data in the dose-escalation cohort, dose levels for PK-expansion were selected, with a total of 8–12 patients enrolled in each level. All patients received a single dose of oral fuzuloparib on day 1 and a single dose of oral apatinib on day 4 in cycle 0 (6 days in total), followed by continuous dosing of fuzuloparib and apatinib in 28-day cycles starting on cycle 1 day 1. Treatment continued until disease progression, intolerable toxicity, or patient withdrawal.
Outcomes
The primary endpoint was to determine the recommended phase 2 dose (RP2D) and tolerability of the fuzuloparib-apatinib combination. Secondary endpoints included best overall response, objective response rate (ORR) and disease control rate (DCR) per RECIST v1.1, cancer antigen 125 (CA-125) response, PFS, overall survival, and PK parameters.
Safety was monitored using AEs, laboratory tests and clinical examinations. AEs were graded according to CTCAE v4.03. DLTs were assessed from the first dose of study treatment (cycle 0 day 1) to the end of cycle 1 of the combination therapy (34 days in total), and defined as any of the following treatment-related AEs (TRAEs): non-hematologic toxicities of grade 3 or worse (with the exception of well-managed grade 3 nausea, vomiting, or diarrhea, and grade 3 creatinine or electrolyte disorder which resolved to grade 1 or baseline level within 24 h), uncontrolled hypertension of grade 3 or worse, grade 4 neutropenia or grade 3 neutropenia accompanied by fever (≥ 38.5 °C), grade 4 thrombocytopenia and toxic effects resulting in dose delay for ≥ 14 days.
Tumor response was assessed using CT or MRI at baseline and every 2 cycles per RECIST v1.1. A complete response (CR) or partial response (PR) was confirmed with a subsequent scan at least 4 weeks after the initial documentation. Disease evaluations based on CA-125 were performed according to the Gynecologic Cancer Intergroup (GCIG) criteria. CA-125 response, defined as a reduction of ≥ 50% in CA-125 level, was assessed in patients with CA-125 levels ≥ 2 folds of the normal upper limit at baseline; the response was confirmed by repeat testing at least 4 weeks apart.
Pharmacokinetic sampling for single dose of fuzuloparib and apatinib was done on day 1 and day 4 (pre-dose and 0.5, 1, 2, 3, 4, 8, 12, 24, 48 h post-dose) of cycle 0, respectively; sampling for continuous dosing of the combination therapy was done on days 1 (pre-dose and 0.5, 1, 2, 3, 4, 8, 12, 24 h post-dose), 8 (pre-dose), 15 (pre-dose), 22 (pre-dose) of cycle 1, and day 1 (pre-dose and 0.5, 1, 2, 3, 4, 8, 12, 24 h post-dose) of cycle 2.
Statistical analysis
All enrolled patients who received at least one dose of study drug were included in the efficacy and safety analysis. Patients who received at least one dose of study drug and were evaluable for DLT (experienced a DLT or completed the whole 34-day assessment period without experiencing a DLT) were included in DLT analysis. Treated patients who had post-dose PK data were included in PK analysis. All efficacy analysis was exploratory in nature. Time-to-event endpoints were estimated using the Kaplan–Meier method, with the 95% CIs calculated using the Brookmeyer and Crowley method. PK parameters were calculated using the WinNonlin noncompartmental model. Safety outcomes were summarized using descriptive statistics. All statistical analyses were done using SAS v9.4.
Results
Patients
Between Mar 17, 2017 and Mar 2, 2021, 52 patients were enrolled: 27 in the dose-escalation cohort and 25 in the PK-expansion cohort. Of them, 30 patients had OC and 22 had TNBC (Fig. 1). At the data cutoff of Aug. 22, 2021, the median follow-up was 11.3 months (IQR 6.6–20.6). The baseline characteristics are summarized in Table 1. Of the 30 patients with OC, 70% had 2 prior lines of treatment and 30% had more than 2 lines. 40% of patients with OC had invasive metastasis, all with multiple metastatic sites; 26.7% had gBRCAmut and 33.3% were platinum-sensitive. Of 22 patients with TNBC, 13.6% had gBRCAmut.

Trial profile
Dose optimization and safety
During dose escalation, 7 dose combinations, up to fuzuloparib 100 mg bid plus apatinib 500 mg qd, were evaluated. Two of 27 patients in the dose-escalation cohort had DLTs in the first cycle. At the dose level of fuzuloparib 100 mg plus apatinib 250 mg, 1 patient experienced febrile neutropenia and grade 4 decreased white blood cell count; at fuzuloparib 100 mg plus apatinib 375 mg, 1 patient experienced grade 4 decreased platelet count. The maximum tolerated dose (MTD) was not reached.
Overall, 19 (36.5%) of 52 enrolled patients had at least one grade ≥ 3 TRAE, with the most common being hypertension (10 [19.2%] patients), anaemia (7 [13.5%]), and decreased platelet count (3 [5.8%]; Table 2). No patients discontinued any component of study treatment due to TRAE. Eleven (21.2%) patients required a dose reduction or interruption. Serious TRAEs were reported in 6 (11.5%) patients, with the most common being decreased platelet count (2 [3.8%] patients). One death (cerebral hemorrhage/brain herniation) in the fuzuloparib 60 mg plus apatinib 250 mg cohort was considered possibly related to study treatment.
PK
The PK profiles of fuzuloparib plus apatinib after a single dose and at steady state are shown in Additional file 1: Table S1 and S2 respectively. At the tested dose levels, plasma exposure of fuzuloparib (AUC0-12 h) and apatinib (AUC0-∞) increased in a dose-dependent manner after a single administration of each agent. After a single co-administration (cycle 1 day 1) of the combination therapy, the plasma exposure of fuzuloparib (AUC0-12 h) appeared unaffected by apatinib dosing, whereas the exposure of apatinib (AUC0-24 h) was reduced by fuzuloparib dosing (by 15.5%-38.3% across dose levels). After continuous dosing of apatinib in combination with fuzuloparib, the AUC0-24 h of apatinib was reduced by 47.7%-65.0% across dose levels (cycle 2 day 1), compared with the corresponding AUC0-∞ after a single dosing of apatinib monotherapy (cycle 0 day 4). The steady-state Cmax and AUC0-24 h of apatinib increased approximately proportionally with increasing doses (from 250 to 500 mg), when administrated with fuzuloparib at a dose of 100 mg bid.
Clinical activity
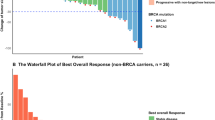
Change in size of target lesion from baseline for each patient is shown in Fig. 2. Across all dose levels, 5 (9.6%) of 52 patients had CR, 14 (26.9%) had PR, and 15 (28.8%) had stable disease (SD) per RECIST v1.1. The confirmed ORR and DCR in all patients were 36.5% (19/52; 95% CI 23.6–51.0) and 65.4% (34/52; 95% CI 50.9–78.0) respectively. At the highest dose level of fuzuloparib 100 mg plus apatinib 500 mg, the confirmed ORR and DCR were 50.0% (4/8; 95% CI 15.7–84.3) and 62.5% (5/8; 95% CI 24.5–91.5) respectively. Taken together with the general good tolerability and dose-proportional exposure of the combination therapy within tested dose levels, fuzuloparib 100 mg bid plus apatinib 500 mg qd was determined to be the RP2D.

Waterfall plot of the best response in target lesion. Tumor response was assessed by the investigator according to RECIST version 1.1. OC, ovarian cancer; TNBC, triple-negative breast cancer
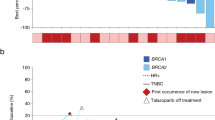
By tumor type, 8 of 10 (80%; 4 CR) patients with platinum-sensitive OC and 6 of 17 (35.3%; 1 CR) patients with platinum-resistant OC achieved confirmed objective response; additional 3 (17.6%) patients with platinum-resistant disease had unconfirmed PR. One (10%) patient with platinum-sensitive OC and 7 (41.2%) patients with platinum-resistant OC had SD, resulting in a DCR of 90.0% (9/10) for platinum-sensitive OC and 76.5% (13/17) for platinum-resistant OC. In responders, median DoR was 9.3 months (95% CI 7.4-not reached [NR]) in patients with platinum-sensitive OC and 18.4 months (95% CI 12.0-NR) in those with platinum-resistant OC (Table 3). By data cutoff, median PFS was 12.1 months (95% CI 1.9-NR) in patients with platinum-sensitive OC and 5.8 months (95% CI 2.1–18.5) in those with platinum-resistant OC. According to BRCA mutation status, the proportion of OC patients achieving confirmed objective response (62.5% [5/8] vs 40.9% [9/22]) and disease control (87.5% [7/8] vs 68.2% [15/22]) was numerically higher in those with gBRCA1/2mut disease than with gBRCA1/2 wild type (gBRCA1/2wt) disease (Additional file 1: Table S3); the median PFS was 9.4 months (95% CI 1.9–20.5) in patients with gBRCA1/2mut OC and 6.7 months (95% CI 2.0–14.9) in those with gBRCAwt OC (p = 0.3951; Fig. 3A, B). In patients who had platinum-sensitive OC, all 3 patients (100%) with gBRCA1/2mut and 5 of 7 (71.4%) patients with gBRCAwt had an objective response; in patients with platinum-resistant OC, an objective response was observed in 2 of 4 (50.0%) of patients harboring gBRCA1/2mut and in 4 of 13 (30.8%) patients harboring gBRCAwt.

Efficacy by BRCA mutation status. Representative BRCA mutations and PFS by BRCA mutation status in patients with ovarian cancer (A, B) and TNBC (C, D). Among 8 patients with gBRCAmut ovarian cancer, 5 had gBRCA1mut, 2 had gBRCA2mut and 1 had both gBRCA1mut and gBRCA2mut. Among 3 patients with gBRCAmut TNBC, all had gBRCA1mut. gBRCAmut, germline BRCA mutation; gBRCAwt, wild-type germline BRCA; PFS, progression-free survival; TNBC, triple-negative breast cancer
In 22 patients with TNBC, 0 CR, 5 (22.7%) confirmed PR and 7 (31.8%) SD were observed and the DCR was 54.5% (12/22; Table 3). In 3 patients harboring gBRCAmut, 2 (66.7%) PR and 1 (33.3%) SD were recorded. By data cutoff, the median PFS were 5.5 months (95% CI 3.8-NR) in patients with gBRCAmut TNBC and 2.8 months (95% CI 1.9–5.8) in those with gBRCAwt TNBC (p = 0.6825; Fig. 3C, D).
Discussion
In this phase 1 trial, the chemo-free combination of fuzuloparib and apatinib demonstrated good tolerability and therapeutic efficacy in biomarker-unselected patients with recurrent gynecological cancers. No MTD was established, and no saturation of plasma drug exposure was observed up to fuzuloparib 100 mg bid plus apatinib 500 mg qd.This highest tested dose combination was determined to be the RP2D. The safety profile of the fuzuloparib-apatinib combination was consistent with the individual agents, with no new safety signals identified. The most frequently reported grade 3 or worse TRAEs were hypertension (the most common TRAE associated with apatinib monotherapy [21, 22, 25]) and hematologic toxicities (the most common TRAEs associated with fuzuloparib monotherapy [17,18,19]). These events were generally manageable with standard supportive care and dose modification, without leading to treatment discontinuation.
In this study, obvious anti-tumor activity was seen for fuzuloparib plus apatinib, with a confirmed ORR of 36.5% and DCR of 65.4% across all dose levels. Specifically, the ORR was 80% for patients with platinum-sensitive recurrent OC, which was much higher than the 35.3% for patients with platinum-resistant recurrent OC. The cross-sensitivity between platinum-based chemotherapy and fuzuloparib-apatinib combination could be partly explained by the overlap between resistance mechanisms of platinum and PARP inhibitor, which involves the reactivation of the HR repair pathway [26]. Notably, the gBRCA1/2 mutation rate of 26.7% for HGSOC patients in our study was slightly higher than the reported rates of 19–23.8% for BRCA-unselected OC patients (> 90% serous histology) in phase 3 trials of other PARP inhibitors [27,28,29]. This difference could be attributed to the relatively small sample size of this study. Consistent with previous reports [12, 13, 29], BRCA1/2mut was predictive of favorable efficacy outcome in OC and the ORR ranged from 30.8% in gBRCAwt, platinum-resistant disease to 100% in gBRCAmut, platinum-sensitive disease in this study. In a previous phase 2 trial, fuzuloparib monotherapy achieved an ORR of 69.9% and a median PFS of 12.0 months in patients with gBRCAmut, platinum-sensitive OC (~ 70% received 2 prior lines of chemotherapy and none received PARP inhibitor) [17]. In this study, our subjects represented a heavily pretreated patient population with recurrent OC, similar to that in the phase 2 trial of fuzuloparib monotherapy. Although the sample size was limited, the ORR and PFS achieved with the combination therapy in patients with platinum-sensitive OC with or without gBRCAmut, appeared to be comparable to those achieved with fuzuloparib monotherapy in patients with platinum-sensitive OC with gBRCAmut. Given the established superior efficacy of PARP inhibitors in treating patients with gBRCAmut OC [13, 29], our preliminary data support the potential clinical benefits with the addition of apatinib.
A handful of clinical trials have assessed the combination of an anti-angiogenic agent and a PARP inhibitor as treatment for recurrent OC, and the ORR ranged from 60%-79.6% in platinum-sensitive disease [11,12,13, 29] and 11.1%-15.4% in platinum-resistant disease [14,15,16]. Direct comparison between studies was difficult considering the differences in patients characteristics (e.g. proportion of patients with BRCA mutation/HR deficiency) and study design (e.g. no. of lines of prior treatment [chemotherapy, PARP inhibitor, and anti-angiogenic agent] allowed); nevertheless, the ORR of 30.8% with fuzuloparib-apatinib in patients with gBRCAwt, platinum-resistant OC was encouraging for a difficult-to-treat population. Importantly, tumor response with fuzuloparib plus apatinib in OC was durable regardless of platinum sensitivity or BRCA mutation status, which highlights the necessity for developing new biomarker to better identify potential responders in the gBRCAwt, platinum-resistant OC population. Notably, in a recent phase 3 trial, cediranib plus olaparib failed to significantly extend PFS versus standard chemotherapy (median, 10.4 vs 10.3 months) in patients with platinum-sensitive OC (64.6% with 1 line of prior therapy). This was speculated to be attributed to the decreased dosing intensity (71.6% of participants required dose modification) and increased rate of treatment discontinuation (21.2% withdrew treatment) due to AEs with the combination [29]. In this study, treatment with fuzuloparib plus apatinib resulted in a median PFS of 12.1 months in patients with platinum-sensitive OC progressing after ≥ 2 lines of platinum-based chemotherapy. The combination was well tolerated, with only 21.2% requiring dose modification and none requiring dose discontinuation due to TRAE. The overall favorable safety profile of fuzuloparib-apatinib supports long-term and sustained use of these drugs, which potentially distinguishing this combination from others under development.
Although the PARP inhibitors olaparib and talazoparib have shown robust efficacy in gBRCAmut TNBC, the prevalence of this mutation is low, ranging from 10–20% in TNBC [30,31,32]. A phase 1 trial has evaluated the combination of olaparib with cediranib in pretreated TNBC patients with or without BRCA1/2 mutation; however, the trial observed limited anti-tumor activity (no objective response and 2 SD in 8 patients), likely due to the small sample size. In the present study, fuzuloparib plus apatinib resulted in an ORR of 22.7% and DoR of 4.5 months in patients with TNBC who had received up to 2 prior lines of chemotherapy. While the clinical activity was modest, the fuzuloparib-apatinib combination as a later-line therapy showed similar effectiveness to conventional chemotherapy and had fewer toxicities in treating a general TNBC population [32, 33], suggesting that it might be considered as chemo-free option under specific conditions. Alternatively, an encouraging ORR of 66.6% and DCR of 100% was obtained with the combination in the subgroup of gBRCA1/2mut TNBC. Based on the consistent efficacy of PARP inhibitors shown in gBRCA1/2mut, HER2- BC, and the promising results observed in phase 1 trials for fuzuloparib with or without apatinib in gBRCA1/2mut BC [19], we have initiated a phase 1/3 trial (NCT04296370) to assess fuzuloparib with or without apatinib, compared to investigator's choice of chemotherapy, in the treatment of gBRCAmut, HER2- BC. This ongoing study includes a run-in phase 1 part, followed by a 3-arm, randomized, phase 3 part. The trial will provide definitive evidence regarding the impact of including apatinib in the treatment regimen for HER2- BC.
When used as combination therapy, the PK profile of fuzuloparib (40–100 mg bid) was similar to that of fuzuloparib monotherapy; however, the exposure of apatinib (250–500 mg qd) at steady state was reduced by 47.7%-65.0% across dose levels when compared to apatinib used as monotherapy. This observation indicates the presence of drug-drug interaction. As the PK profiles of two drugs in multiple doses for combination therapy have been investigated, the data imply that higher dose level of apatinib may be tolerable when combined with fuzuloparib. Besides, the decreased exposure of apatinib suggests potential lower related toxicity of apatinib when used in combination with fuzuloparib. Based on the PK data, it’s recommended that the dose of apatinib be adjusted first in the combination, in the management of intolerable AEs that were potentially related to either agent.
Conclusions
In summary, the combination of fuzuloparib and apatinib showed hematologic DLTs and acceptable safety in patients with recurrent OC and TNBC. Fuzuloparib 100 mg bid plus apatinib 500 mg qd was determined to be the RP2D. With the activity observed in OC and TNBC, further investigation of the combination therapy is warranted as a potential alternative to cytotoxic treatment.
Availability of data and materials
Datasets supporting the conclusions of the study are presented in the paper or are available from the corresponding authors upon reasonable request.
Abbreviations
- AE:
-
Adverse event
- bid:
-
Twice daily
- CA-125:
-
Cancer antigen 125
- CR:
-
Complete response
- CTCAE:
-
Common Terminology Criteria for Adverse Events
- DCR:
-
Disease control rate
- DLT:
-
Dose-limiting toxicity
- ECOG:
-
Eastern Cooperative Oncology Group
- gBRCA :
-
Germline BRCA
- GCIG:
-
Gynecologic Cancer Intergroup
- HGSOC:
-
High-grade serous ovarian cancer
- HR:
-
Homologous recombination
- MTD:
-
Maximum tolerated dose
- ORR:
-
Objective response rate
- PARP:
-
Poly (ADP-ribose) polymerase
- PFS:
-
Progression-free survival
- PK:
-
Pharmacokinetics
- PR:
-
Partial response
- qd:
-
Once daily
- RECIST:
-
Response Evaluation Criteria in Solid Tumors
- RP2D:
-
Recommended phase 2 dose
- TNBC:
-
Triple-negative breast cancer
- TRAE:
-
Treatment-related adverse event
- VEGF:
-
Vascular endothelial growth factor
- VEGFR:
-
Vascular endothelial growth factor receptor
References
Network CGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15.
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–21.
O’Connor MJ. Targeting the DNA Damage Response in Cancer. Mol Cell. 2015;60:547–60.
Vanacker H, Harter P, Labidi-Galy SI, Banerjee S, Oaknin A, Lorusso D, et al. PARP-inhibitors in epithelial ovarian cancer: Actual positioning and future expectations. Cancer Treat Rev. 2021;99: 102255.
Mirza MR, Pignata S, Ledermann JA. Latest clinical evidence and further development of PARP inhibitors in ovarian cancer. Ann Oncol. 2018;29:1366–76.
Bianchini G, De Angelis C, Licata L, Gianni L. Treatment landscape of triple-negative breast cancer - expanded options, evolving needs. Nat Rev Clin Oncol. 2022;19:91–113.
Lim JJ, Yang K, Taylor-Harding B, Wiedemeyer WR, Buckanovich RJ. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia. 2014;16:343–53 .e341–2.
Bindra RS, Crosby ME, Glazer PM. Regulation of DNA repair in hypoxic cancer cells. Cancer Metastasis Rev. 2007;26:249–60.
Bindra RS, Gibson SL, Meng A, Westermark U, Jasin M, Pierce AJ, et al. Hypoxia-induced down-regulation of BRCA1 expression by E2Fs. Cancer Res. 2005;65:11597–604.
Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann Oncol. 2019;30:551–7.
Mirza MR, Åvall Lundqvist E, Birrer MJ, dePont CR, Nyvang GB, Malander S, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol. 2019;20:1409–19.
Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, et al. Combination cediranib and olaparib versus olaparib alone for women with recurrent platinum-sensitive ovarian cancer: a randomised phase 2 study. Lancet Oncol. 2014;15:1207–14.
Colombo N, Tomao F, Benedetti Panici P, Nicoletto MO, Tognon G, Bologna A, et al. Randomized phase II trial of weekly paclitaxel vs. cediranib-olaparib (continuous or intermittent schedule) in platinum-resistant high-grade epithelial ovarian cancer. Gynecol Oncol. 2022;164:505–13.
Nicum S, Holmes J, McGregor N, Dunn R, Collins L, Kaye S, et al. Randomised phase II trial of olaparib compared to weekly paclitaxel or olaparib plus cediranib in patients with platinum-resistant ovarian cancer (OCTOVA). Ann Oncol. 2021;32:S725–6.
Lee JM, Moore RG, Ghamande S, Park MS, Diaz JP, Chapman J, et al. Cediranib in combination with olaparib in patients without a germline BRCA1/2 mutation and with recurrent platinum-resistant ovarian cancer: phase IIb CONCERTO trial. Clin Cancer Res. 2022;28:4186–93.
Li N, Bu H, Liu J, Zhu J, Zhou Q, Wang L, et al. An open-label, multicenter, single-arm, phase II study of fluzoparib in patients with germline BRCA1/2 mutation and platinum-sensitive recurrent ovarian cancer. Clin Cancer Res. 2021;27:2452–8.
Li N, Zhang Y, Wang J, Zhu J, Wang L, Wu X, et al. Fuzuloparib maintenance therapy in patients with platinum-sensitive, recurrent ovarian carcinoma (FZOCUS-2): a multicenter, randomized, double-blind, placebo-controlled, phase III trial. J Clin Oncol. 2022;40:2436–46.
Li H, Liu R, Shao B, Ran R, Song G, Wang K, et al. Phase I dose-escalation and expansion study of PARP inhibitor, fluzoparib (SHR3162), in patients with advanced solid tumors. Chin J Cancer Res. 2020;32:370–82.
Wang L, Yang C, Xie C, Jiang J, Gao M, Fu L, et al. Pharmacologic characterization of fluzoparib, a novel poly(ADP-ribose) polymerase inhibitor undergoing clinical trials. Cancer Sci. 2019;110:1064–75.
Li J, Qin S, Xu J, Xiong J, Wu C, Bai Y, et al. Randomized, double-blind, placebo-controlled phase III trial of apatinib in patients with chemotherapy-refractory advanced or metastatic adenocarcinoma of the stomach or gastroesophageal junction. J Clin Oncol. 2016;34:1448–54.
Qin S, Li Q, Gu S, Chen X, Lin L, Wang Z, et al. Apatinib as second-line or later therapy in patients with advanced hepatocellular carcinoma (AHELP): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2021;6:559–68.
Hu N, Zhu A, Si Y, Yue J, Wang X, Wang J, et al. A phase II, single-arm study of apatinib and oral etoposide in heavily pre-treated metastatic breast cancer. Front Oncol. 2020;10: 565384.
Lan C-Y, Wang Y, Xiong Y, Li J-D, Shen J-X, Li Y-F, et al. Apatinib combined with oral etoposide in patients with platinum-resistant or platinum-refractory ovarian cancer (AEROC): a phase 2, single-arm, prospective study. Lancet Oncol. 2018;19:1239–46.
Li H, Geng C, Zhao H, Jiang H, Song G, Zhang J, et al. Multicenter phase II study of apatinib single or combination therapy in HER2-negative breast cancer involving chest wall metastasis. Chin J Cancer Res. 2021;33:243–55.
Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 2015;5:1137–54.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390:1949–61.
Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381:2403–15.
Liu JF, Brady MF, Matulonis UA, Miller A, Kohn EC, Swisher EM, et al. Olaparib with or without cediranib versus platinum-based chemotherapy in recurrent platinum-sensitive ovarian cancer (NRG-GY004): a randomized, open-label, phase III trial. J Clin Oncol. 2022;40:2138–47.
Gonzalez-Angulo AM, Timms KM, Liu S, Chen H, Litton JK, Potter J, et al. Incidence and outcome of BRCA mutations in unselected patients with triple receptor-negative breast cancer. Clin Cancer Res. 2011;17:1082–9.
Engel C, Rhiem K, Hahnen E, Loibl S, Weber KE, Seiler S, et al. Prevalence of pathogenic BRCA1/2 germline mutations among 802 women with unilateral triple-negative breast cancer without family cancer history. BMC Cancer. 2018;18:265.
Robson M, Im S-A, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. 2017;377:523–33.
Bardia A, Hurvitz SA, Tolaney SM, Loirat D, Punie K, Oliveira M, et al. Sacituzumab Govitecan in Metastatic Triple-Negative Breast Cancer. N Engl J Med. 2021;384:1529–41.
Acknowledgements
We are grateful to all patients and their families and all members of the collaborative group in this trial. Medical writing assistance was provided by Xiuzhi Wu, PhD (Jiangsu Hengrui Pharmaceuticals) according to Good Publication Practice Guidelines.
Funding
This study was funded by Jiangsu Hengrui Pharmaceuticals.
Author information
Authors and Affiliations
Contributions
Conceptualization: HL and YG; Investigation: YL, WW, RY, YZ1, YZ2, KZ, HP, KW, GL1, GL2, RZ, KL, YG and HL; Formal analysis: BZ and JR; Project administration: YW; Supervision: QW; Roles/Writing—original draft: HL, YL, WW and RY; Writing—review & editing: all authors. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The trial protocol was approved by the institutional review board or ethnic review committee of all participating centers: Peking University Cancer Hospital and Institute (2017YW02; 2017YW03); West China Second University Hospital, Sichuan University (Y2018009); Qilu Hospital of Shangdong University (2018007); Xiangya Hospital, Central South University (201710116); Hunan Cancer Hospital (2018 [No.193]); Sir Run Run Shaw Hospital, Zhejiang University School of Medicine (20180206–2); Tianjin Medical University Cancer Institute and Hospital (E2018053); Harbin Medical University Cancer Hospital (2018–129); Union Hospital, Tongji Medical College, Huazhong University of Science and Technology (2018 [No.19]). All procedures were conducted in accordance with the guidelines of Good Clinical Practice and the Declaration of Helsinki. All patients provided written informed consent before enrollment.
Consent for publication
Not applicable.
Competing interests
JR, BZ, YW and QW were employees of Jiangsu Hengrui Pharmaceuticals at the time of study. All other authors have no conflicts of interest to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Table S1. Pharmacokinetic parameters of fuzuloparib and apatinib after single dosing. Table S2. Pharmacokinetic parameters of fuzuloparib and apatinib after multiple dosing (cycle 2 day 1). Table S3. Tumor response in patients with ovarian cancer by germline BRCA mutation status.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Y., Wang, W., Yin, R. et al. A phase 1 trial of fuzuloparib in combination with apatinib for advanced ovarian and triple-negative breast cancer: efficacy, safety, pharmacokinetics and germline BRCA mutation analysis. BMC Med 21, 376 (2023). https://doi.org/10.1186/s12916-023-03046-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-023-03046-8