
Abstract
Background
Epidemiological and clinical studies have suggested comorbidity between amyotrophic lateral sclerosis (ALS) and autoimmune disorders. However, little is known about their shared genetic architecture.
Methods
To examine the relation between ALS and 10 autoimmune diseases, including asthma, celiac disease (CeD), Crohn’s disease (CD), inflammatory bowel disease (IBD), multiple sclerosis (MS), psoriasis, rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), type 1 diabetes (T1D), and ulcerative colitis (UC), and identify shared risk loci, we first estimated the genetic correlation using summary statistics from genome-wide association studies, and then analyzed the genetic enrichment leveraging the conditional false discovery rate statistical method.
Results
We identified a significant positive genetic correlation between ALS and CeD, MS, RA, and SLE, as well as a significant negative genetic correlation between ALS and IBD, UC, and CD. Robust genetic enrichment was observed between ALS and CeD and MS, and moderate enrichment was found between ALS and UC and T1D. Thirteen shared genetic loci were identified, among which five were suggestively significant in another ALS GWAS, namely rs3828599 (GPX3), rs3849943 (C9orf72), rs7154847 (G2E3), rs6571361 (SCFD1), and rs9903355 (GGNBP2). By integrating cis-expression quantitative trait loci analyses in Braineac and GTEx, we further identified GGNBP2, ATXN3, and SLC9A8 as novel ALS risk genes. Functional enrichment analysis indicated that the shared risk genes were involved in four pathways including membrane trafficking, vesicle-mediated transport, ER to Golgi anterograde transport, and transport to the Golgi and subsequent modification.
Conclusions
Our findings demonstrate a specific genetic correlation between ALS and autoimmune diseases and identify shared risk loci, including three novel ALS risk genes. These results provide a better understanding for the pleiotropy of ALS and have implications for future therapeutic trials.
Similar content being viewed by others

Background
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder, characterized by the death of motor neurons as well as paralysis of voluntary muscles [1]. The majority of ALS patients die within 3~5 years after diagnosis, mostly due to respiratory failure [2]. The pathological mechanisms underlying ALS are multifarious and complex, with a sophisticated interaction between genetic and environmental factors [3]. To date, no effective therapies for ALS have been found, and approved drugs only improved survival to a limited extent [4]. Therefore, exploring the pathogenesis of ALS and developing novel therapeutic strategies are necessary and urgent.
Currently, compelling evidence implicates that dysregulated immunities are associated with ALS [5]. It has been established that neuroinflammation is overactivated in ALS, accompanied by microglia transformation, astrocyte proliferation, perivascular infiltration of monocytes and T cells, and dysregulated immune-related genes [6,7,8,9]. Typical hallmarks of autoimmunity occurring in the pathogenesis of ALS have also been reported, such as the presence of circulating immune complexes and the evidence of higher frequency of specific histocompatibility types [10]. Moreover, epidemiologic studies presented that several pre-existing autoimmune disorders are associated with an increased risk of ALS [11]. And intermediate alleles of C9orf72, the most common genetic cause of ALS, were suggested to be associated with systemic autoimmune diseases, indicating the role of C9orf72 in immunity regulation [12]. Furthermore, mice harboring loss-of-function mutations in the ortholog of C9orf72 cause fatal autoimmune diseases [13]. These associations raise the possibility of shared genetic or environmental risk factors, or clues to modifiable triggers that might thereby affect ALS incidence. Therefore, a systematic study is necessary to decipher whether shared polygenic risk variants exist between ALS and autoimmune diseases, and whether specific molecular biological pathways are involved.
Recently, a novel statistical method to investigate genetic overlapping between polygenic traits using summary data from genome-wide association studies (GWAS) have been developed and utilized extensively in several human traits and diseases [14,15,16]. By incorporating GWAS results from multiple disorders and phenotypes, this method could provide insights into the genetic pleiotropy (defined as a single gene or variant being associated with more than one distinct phenotype) and increased statistical power to discover less significant associations [14,15,16]. Applying this approach, we systematically evaluated the shared genetic risk between ALS and autoimmune diseases and further conducted functional enrichment analysis.
Methods
GWAS summary statistics
We investigated the genetic links between ALS [17] and 10 autoimmune disorders including asthma [18], multiple sclerosis (MS) [19], psoriasis [20], rheumatoid arthritis (RA) [21], systemic lupus erythematosus (SLE) [22], type 1 diabetes (T1D) [23], celiac disease (CeD) [24], inflammatory bowel disease (IBD) [25], Crohn’s disease (CD) [25], and ulcerative colitis (UC) [25] based on GWAS summary statistics. Details of the summary data for all GWAS were shown in Additional file 1: Table S1. The study design including the collection of samples, quality control procedures, and imputation methods have been described in each publication. To confirm the findings in the discovery phase, we further assessed the P values of the identified pleiotropic single-nucleotide polymorphisms (SNP) in another ALS GWAS [26]. The relevant institutional review boards or ethics committees approved the research protocol of each GWAS, and all human participants gave written informed consent.
Statistical analyses
Genetic correlation
We estimated the genetic correlation between ALS and each autoimmune disorder using GNOVA [27]. GNOVA estimates genetic covariance with summary data of the genetic variants shared between two GWAS, and then calculates the genetic correlation based on genetic covariance and variant-based heritability. We ran GNOVA on SNPs in both diseases together with reference data derived from the 1000 Genomes Project European population using default parameters. We did not correct for sample overlap when running GNOVA, since no information was available to evaluate the extent of sample overlap between different GWAS. Additionally, considering that different genomic regions disproportionately contributed to the genome-wide correlation, we further quantified the correlation between ALS and each autoimmune disorder in small regions in the genome using ρ-HESS with default parameters [28].
Genomic control
Due to population stratification or cryptic relatedness or overcorrection of test statistics [29], the empirical null distribution in GWAS is sometimes inflated or deflated. To correct for such bias, we applied a genomic control method leveraging intergenic SNPs to adjust the summary statistics for each GWAS respectively (Additional file 2) [29,30,31,32,33,34]. Then, we pruned the SNPs by removing SNPs in linkage disequilibrium (LD) (r2 > 0.2 within 250 kb) based on 1000 Genomes Project LD structure using plink –clump functionality [34].
Pleiotropic enrichment plots
To assess the pleiotropic enrichment, we plotted conditional quantile-quantile plots for ALS by creating subsets of SNPs based on their association with each autoimmune disorder. To further quantitatively assess the level of enrichment, we constructed fold-enrichment plots of nominal -log10(P) values of ALS for all SNPs and subsets of SNPs determined by the significance of their association with each autoimmune disease (Additional file 2) [14,15,16].
Identification of risk loci
To identify risk loci associated with ALS conditional on each autoimmune disease, we computed the conditional false discovery rate (FDR) statistics using the conditional FDR approach (Additional file 2) [14,15,16]. To reduce false positives, a significance threshold of FDR < 0.01 was utilized. Furthermore, to identify shared risk loci associated with ALS and each autoimmune disease, we computed the conjunctional FDR statistics (Additional file 2) [32, 35, 36]. A significance threshold of FDR < 0.05 was utilized, corresponding to five false positives per 100 reported associations. A stricter threshold was chosen for the conditional statistics since its possibility of false positive was greater. Then, we built the conditional and conjunctional Manhattan plots to illustrate the identified risk loci between ALS and each autoimmune disease (Additional file 2) [14,15,16]. The annotated gene for each significant locus by ANNOVAR was listed in the plots [33]. For intergenic variants spanning more than 1 gene, the significant variant in each gene will be listed. We used the R implementation of the conditional FDR method available from github.com/KehaoWu/GWAScFDR.
Functional evaluation of shared risk loci
To assess whether the shared risk loci modify gene expression, we evaluated cis-expression quantitative trait loci (eQTL) in Braineac, a publicly available dataset of normal control brains for investigating the genes and SNPs associated with neurological disorders [37]. We analyzed eQTL for the mean P value derived across these brain regions: the cerebellum, frontal cortex, hippocampus, medulla, occipital cortex, putamen, substantia nigra, temporal cortex, thalamus, and white matter. To minimize false positives, a P value below 1.0E−04 was considered as significant after Bonferroni correction. Meanwhile, we also analyzed cis-eQTL in whole blood, skeletal muscle, and 13 brain tissues (amygdala, anterior cingulate cortex (BA24), caudate basal ganglia, cerebellar hemisphere, cerebellum, cortex, frontal cortex (BA9), hippocampus, hypothalamus, nucleus accumbens basal ganglia, putamen basal ganglia, spinal cord cervical, and substantia nigra) from GTEx v7 [38]. Cis-eQTLs as pre-computed by GTEx were downloaded directly from the GTEx portal (http://gtexportal.org/). We applied a nominal P value cutoff of 1E−06 to identify significant cis-eQTLs, which approximates a false discovery threshold of 0.05.
To identify enrichments in gene ontologic features associated with ALS and autoimmune disorders, we used ConsensusPathDB [39] for functional interaction analysis. The shared risk genes identified with the conjunctional FDR method and eQTL analyses were utilized with default parameters and background gene sets. Biological, cellular, and molecular gene ontologic terms were analyzed. Genes in the HLA region were excluded due to the complex LD patterns.
Results
Genetic correlation
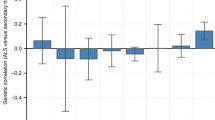
We identified a significant positive genetic correlation between ALS and CeD, MS, RA, and SLE, as well as a significant negative genetic correlation between ALS and IBD, UC, and CD (Additional file 1: Table S2). To serve as comparison, we also explored the correlation between Parkinson’s disease (PD) [40], Alzheimer’s disease (AD) [41], and autoimmune diseases using GNOVA with the same pipeline. As a result, significant correlation was only identified between PD and RA after the Bonferroni correction (Additional file 1: Table S2). These results suggested the considerable genetic links between ALS and autoimmune disorders. Furthermore, we searched for genomic regions that disproportionately contributed to the genetic correlation. No regions with significant correlation were identified after Bonferroni correction, while regions with nominally significant correlation were found between ALS and MS, IBD, SLE, CD, UC, and RA (Additional file 1: Figure S1).
Estimation of pleiotropic enrichment
In the stratified quantile-quantile plots for ALS conditional on association P values with each autoimmune disease, successive enrichment was found for CeD and MS, and moderate enrichment was found for UC and T1D (Fig. 1), indicating that the proportion of non-null SNPs in ALS increased with higher levels of association with these diseases. In contrast, minimal or no enrichment was found for the other diseases. In the fold-enrichment plots, we could observe over 300-fold enrichment conditional on CeD, approximately 22-fold enrichment on MS, 9-fold enrichment on UC, and 5-fold enrichment on T1D for progressively stringent P value thresholds, while minimal enrichment on the other diseases, suggesting a selective genetic overlap between ALS and autoimmune disorders (Fig. 2).

Enrichment plots. Conditional quantile-quantile plots of nominal versus empirical -log10(P) of ALS as a function of significance of association with autoimmune diseases. Dashed lines indicate the null hypothesis. The figure is suggested to be viewed online for higher resolution

Fold-enrichment plots of nominal -log10(P) of ALS as a function of significance of association with autoimmune diseases. The figure is suggested to be viewed online for higher resolution
ALS-associated loci identified with conditional FDR
To discover genetic variants associated with ALS conditional on each autoimmune disease, we performed the conditional FDR statistical analysis. A total of 32 risk loci were identified with conditional FDR < 0.01 (Additional file 1: Table S3, Fig. S2), including 23 novel loci which were not significant (P < 5.0E−08) in the original ALS GWAS. Among these 32 loci, 11 were suggestively significant (P < 1.0E−05) in the replication GWAS, namely rs3828599 (GPX3), rs10463311 (TNIP1), rs17070492 (LOC101927815), rs9969832 (MOB3B), rs2484319 (C9orf72), rs12308116 (C12orf56), rs447614 (G2E3), rs1950882 (SCFD1), rs35714695 (SARM1), rs2285642 (GGNBP2), and rs12608932 (UNC13A) (Additional file 1: Table S3). Among these replicated genes, GPX3, TNIP1, MOB3B, C9orf72, SCFD1, SARM1, and UNC13A have been described as risk genes for ALS by earlier GWAS, while the others were novel risk genes, including C12orf56, G2E3, TRIP11, and GGNBP2.
Risk loci shared between ALS and autoimmune diseases
To identify shared loci between ALS and autoimmune diseases, we further calculated the conjunctional FDR statistics. A total of 13 shared risk loci were identified with conjunctional FDR < 0.05 (Table 1, Fig. 3, Additional file 1: Figure S3). Among the 13 loci, 5 were suggestively significant (P < 1.0E−05) in the replication ALS GWAS, namely rs3828599 (GPX3), rs3849943 (C9orf72), rs7154847 (G2E3), rs6571361 (SCFD1), and rs9903355 (GGNBP2). Among the 5 genes, G2E3 and GGNBP2 were newly discovered risk genes for ALS (Table 1). No shared genetic loci were found between ALS and asthma, psoriasis, RA, and SLE, which was consistent with the stratified QQ plots and fold-enrichment plots with no apparent enrichment observed.

Conjunctional Manhattan plots showing shared genetic loci between ALS and each autoimmune disease. The red horizontal line represents the significant threshold (conjunctional FDR < 0.05)
Functional interpretation of shared risk loci
To determine the functional effects of these shared risk loci, we evaluated cis-eQTL in Braineac and GTEx. As a result, the pleiotropic risk loci affect the expression of GGNBP2, ATXN3, and SLC9A8 in tissues from both Braineac and GTEx. In addition, the pleiotropic risk loci affect the expression of DCTN4 and TEK in brain regions from Braineac (Table 2), and TRIP11, NOD2, SCFD1, C9orf72, DENND6B, PLXNB2, PPP6R2, DHRS11, MYO19, ZNHIT3, and TMEM116 in tissues from GTEx (Additional file 1: Table S4).
To determine the biological pathways represented by shared risk genes and the genes identified with cis-eQTL analyses, we conducted pathway over-representation analysis. Four pathways were enriched, namely ER to Golgi anterograde transport (P = 5.70E−03), membrane trafficking (P = 1.02E−03), transport to the Golgi and subsequent modification (P = 8.47E−03), and vesicle-mediated transport (P = 1.29E−03). Additionally, 6 GO sets were identified (Additional file 1: Table S5).
Discussion
In the current study, we investigated the pleiotropy between ALS and autoimmune disorders using summary statistics from large GWAS and the conditional FDR statistical method. We identified significant genetic enrichment for ALS as a function of CeD and MS and moderate enrichment of UC and T1D. Besides, we identified 13 significant shared loci, with 5 validated in the replication ALS GWAS, and 3 were annotated as related to gene expression in tissues from both Braineac and GTEx. These results clarified the shared genetic architecture between ALS and autoimmune diseases, suggested that ALS pathogenesis might be mediated by the dysfunctioning of the immune system, and provided a better understanding for the pleiotropy of ALS.
Neurodegenerative disorders are complex diseases characterized by loss of neurons and axons in the central nervous system. Recently, there is increasing recognition that inflammation and immune response play important roles in the pathogenesis of neurodegeneration [42]. Activated microglia, which is a key regulator of brain inflammation, is also an important cause for neuronal loss in models of neurodegenerative diseases [43]. Previous studies have investigated pleiotropy between AD, PD and autoimmune diseases [15, 16], and identified shared risk genes. However, the pleiotropy between ALS and autoimmune disorders is still not clear. Our results supported the hypothesis of shared genetic risk between ALS and autoimmune diseases and supplemented current knowledge for the correlation between neurodegeneration and autoimmunity.
We observed a substantial genetic enrichment between CeD and ALS. CeD is an autoimmune disorder that occurs in genetically predisposed individuals who develop an immune reaction to gluten [44]. Meanwhile, gluten sensitivity has also been shown to induce neurologic manifestations [45]. For example, a recent case-control designed study suggested that ALS might be associated with autoimmunity and gluten sensitivity [44]. However, such links still need to be confirmed further, as subsequent studies reported inconsistent results [46]. We also identified a shared risk gene GPX3, which could have functional relevance to both diseases. GPX3 is an antioxidant molecule functionally related to SOD1 [47], the first causative gene for ALS. In a mass spectrometric screen of sera of SOD1H46R rats compared to wild-type controls, GPX3 expression was increased by 1.3 fold in the pre-symptomatic stage, while decreased by 0.74 fold in the end stage of the disease [48]. Meanwhile, GPX3 activity was reduced in CeD patients (P < 0.001) in a recent pilot study, suggesting its potential role in the pathogenesis of CeD [49]. In contrast, minimal enrichment was observed between ALS and CD, which has partially shared genetic basis and pathogenesis with CeD [50]. However, the strongest signal rs2076756 (NOD2) in the conjunctional Manhattan plot was identified between ALS and CD. NOD2 is a member of the pattern-recognition receptor family and can recognize muramyl dipeptide in cytomembrane to activate the NF-κB pathway and stimulate inflammatory factor response [51]. Recently, NOD2 was identified as a potent autophagy inducer as well, suggesting its potential role in neurodegeneration [52]. In addition, moderate enrichment was observed between ALS and UC, and G2E3-SCFD1 was identified as shared risk genes between ALS and IBD. These results together suggested a potential link between ALS and chronic inflammation of the gastrointestinal tract, though such link was not consistently detected based on current results. Further explorations are warranted to elucidate the correlation.
We also observed genetic enrichment between ALS and MS. Both ALS and MS are complex neurological disorders with sophisticated interactions of environmental toxicity and genetic predisposition [53]. By far, the origin of MS and ALS is still unknown, but the progressive central axonal degeneration is seen in both MS and ALS. And there is evidence suggesting shared cellular mechanisms affecting the disease progression, particularly glial responses in the two disorders [54]. One explanation would be that the two conditions share some common genes which predispose to both MS and ALS. Here, we identified six shared risk genes including TRIP11, ATXN3, GGNBP2, SLC9A8, DENND6B, and ANO5 (Table 1). The six shared genes were enriched in two significant cellular components, namely organelle subcompartment (GO:0031984, P = 0.0008) and endomembrane system (GO:0012505, P = 0.0032) based on results from ConsensusPathDB. Therefore, the membrane system may implicate the overlapping factors related to MS and ALS, and the shared genes might be responsible for the link.
Association between ALS and diabetes has been observed in epidemiological studies [55, 56]. Recently, a causal protective role of type 2 diabetes on ALS was noticed in the European population [57]. In contrast, the association between T1D and ALS was still less understood, and T1D might increase the risk for ALS based on a recent retrospective population-based study [11, 56], though such results were still awaiting further replications [58]. In our study, we observed enrichment between ALS and T1D, and identified three shared risk genes C9orf72, ZNF184, and ATXN2. Repeat expansions in C9orf72 is a frequent cause of ALS, and C9orf72 carriers tend to have autoimmune diseases more frequently, suggesting autoimmune inflammation may be intrinsically linked to ALS pathophysiology [59]. Meanwhile, a recent study found that mutations disrupting the normal function of C9orf72 cause mice to develop features of autoimmunity [12]. Thus, C9orf72 might serve as an important factor related to inflammation and autoimmunity [12]. ZNF184 is in the extended major histocompatibility complex (MHC) region, which plays a complex but important role in both neurodegenerative and autoimmune diseases. ATXN2 has been reported to be associated with several autoimmune diseases like T1D [60], CD [61], and CeD [62] by GWAS. Meanwhile, high-length repeats of CAG trinucleotide in ATXN2 was identified as a risk factor for ALS as well [63, 64]. Taken together, genetic correlation exists between ALS and T1D, and genes such as C9orf72, ZNF184, and ATXN2 might function as potential links between the two diseases.
Compared with CeD, MS, T1D, and UC, we did not detect enrichment between ALS and RA, psoriasis, asthma, CD, and SLE. The results were to a large extent in agreement with a previous epidemiologic study which investigated whether ALS incidence was higher in people with prior autoimmune diseases [11]. This study found that patients with CeD, younger-onset diabetes, MS, asthma, and SLE were at higher risk of ALS, and the rate ratio for UC was borderline significant (P = 0.05), and no significant difference was found for RA, psoriasis and CD. As for the different results for asthma and SLE between our study and this epidemiological study, further explorations were warranted to elucidate their correlation with ALS. Moreover, we noted that both positive and negative genetic correlations were observed between ALS and autoimmune disorders (Additional file 1: Table S2). We identified a significant and positive genetic correlation between ALS and CeD, MS, RA, and SLE, indicating that the direction of effect of risk-increasing and protective alleles is consistently aligned between ALS and these autoimmune disorders at genome wide. In contrast, we found a significant and negative genetic correlation between ALS and IBD, UC, and CD, implying these diseases may possess divergent biological mechanisms from the other autoimmune disorders. Such results were in line with previous epidemiological research, which detected no significant difference in the incidence rate of ALS among patients with prior UC and CD compared with controls [11]. Additionally, IBD, including UC and CD, was characterized by chronic inflammation of the gastrointestinal tract, which fulfilled some of the criteria required for classification as autoimmune disorders, while the extent of the involvement remained to be determined [65]. Taken together, immune and autoimmune mechanisms are complex and may vary from disease to disease, so a deeper investigation into the molecular mechanisms involved in these diseases is necessary to further understand the correlation.
Combining cFDR analyses results and eQTL analyses results from Braineac and GTEx, we identified three risk genes, namely SLC9A8, ATXN3, and GGNBP2. SLC9A8 is a kind of sodium-hydrogen exchangers (NHEs) that exchange extracellular Na+ for intracellular H+. Ionic homeostasis dysregulation has been proposed as the main trigger of the pathological cascade which brings to motor-neuron loss [66]. A recent study also observed a marked increase in the persistent component of the Na+ current from pre-symptomatic SOD1G93A mice [67, 68], suggesting the potential role of SLC9A8 in ALS etiology. ATXN3 provides instructions for making an enzyme called ataxin-3, which is involved in destroying excessive or damaged proteins. A trinucleotide repeat expansion in ATXN3 could cause spinocerebellar ataxia type 3, a neurologic disorder that is characterized by progressive ataxia. Mutations in ATXN3 affect neurons and other types of brain cells and alter transcription of multiple signal transduction pathways including depressed Wnt and elevated growth factor pathways [69]. Moreover, GWAS has identified suggestively significant SNPs in ATXN3 as associated with ALS [17]. These findings suggest the potential involvement of ATXN3 in ALS. Previous studies have suggested the potential role of GGNBP2 in ALS through gene-based association analysis and summary statistics-based Mendelian randomization (SMR) analysis [26, 70]. Meanwhile, we noticed that GGNBP2 is an important tumor suppresser involved in several kinds of cancers [71]. Cancer and neurodegenerative diseases are like the two sides of a coin. The pathways that cause neuronal apoptosis, like mitogen-activated protein kinase (MAPK) signaling, can cause uncontrolled neuronal growth as well. Epidemiological studies have shown that the overall risk of cancer was significantly reduced in patients with ALS [72]. Therefore, GGNBP2 might act as potential genetic links between ALS and cancer.
Using the pleiotropic genes for functional enrichment analysis in ConsensusPathDB, we identified four enriched pathways, all of which were somehow involved in the pathogenesis of ALS. Membrane trafficking has been implicated in virtually every aspect of neuronal function, particularly neuronal maintenance and degeneration [73]. Intracellular membrane trafficking defects affecting key neuronal functions may be an early determinant of motor neuron loss in ALS [74]. Meanwhile, the molecular regulation of intracellular and extracellular vesicle trafficking is an important pathway in ALS pathogenesis [75], and mutation in the vesicle-trafficking protein has been implicated to cause late-onset spinal muscular atrophy and ALS. Fragmentation of the Golgi apparatus has been detected in motor neurons of ALS patients and animal models [76], and mutant SOD1 was shown to inhibit secretory protein transport from the ER to Golgi apparatus [77].
Combining results from previous publications that estimate pleiotropy between AD, PD and autoimmune diseases using the conditional FDR method, we found that all three neurodegenerative disorders were strongly or moderately enriched on immune-mediated enteropathy like CeD, CD, and UC. Gut microbiota has been shown to play an essential role in IBD and CeD [78], while in recent years, gut microbiome alterations were found to be closely related to neurodegeneration as well [79]. The microbiota-gut-brain axis composed of endocrinological, immunological, and neural mediators was even proposed due to its involvement in neurodegenerative diseases [80]. Our pleiotropic analyses together with previous studies further emphasized that gut microbiota plays an important role in the pathogenesis of neurodegenerative disorders. In addition, differential enrichment was also observed between autoimmune disorders and AD, PD, and ALS respectively. The diversity suggested the variation between the pathogenesis of different neurodegenerative disorders.
Strengths and limitations
Using the pleiotropy-based statistical method, we identified novel significant SNPs associated with ALS and autoimmune disorders. These SNPs were mostly suggestively significant in the original studies but overlooked due to the limitation of the current GWAS sample size. Moreover, the current results have clinical implications. Since we combined GWAS summary statistics from different but related diseases, the findings may increase our understanding of the pathogenetic mechanisms influenced by pleiotropic loci and facilitate novel treatment strategies in clinical trials. However, the GWAS used in the current study were mostly performed on participants of European ancestry; thus, the findings of shared genetic architecture might be biased and not applicable to other populations. Meanwhile, there was a potential sample overlap between each GWAS. Such overlap might bring some bias to the statistical analysis, although the bias will be minimal. Meanwhile, there was sample overlap between the discovery ALS GWAS and replication ALS GWAS. To strengthen the validity of the identified pleiotropic SNPs, we utilized a more stringent significance threshold (P = 1E−05) instead of the commonly used threshold in replication (P = 0.05). In addition, the sample size for some diseases like CeD and SLE was relatively smaller, which might influence the results to some extent. Besides, although we identified several pleiotropic SNPs, how these SNPs or genes involved in the pathogenesis of ALS and autoimmune diseases were still little understood. Further functional explorations will provide better understandings.
Conclusions
By integrating GWAS summary data and the conditional FDR statistical method, we identified selective pleiotropy and novel shared loci between ALS and autoimmune diseases. We further identified novel ALS risk genes SLC9A8, ATXN3, and GGNBP2 by combining eQTL analyses. These findings could provide novel insights into the shared genetic background between ALS and autoimmunity and help better understand the etiology of ALS.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Code or algorithm used to generate results in this study is available from the corresponding authors on reasonable request.
Abbreviations
- ALS:
-
Amyotrophic lateral sclerosis
- GWAS:
-
Genome-wide association study
- SNP:
-
Single-nucleotide polymorphism
- LD:
-
Linkage disequilibrium
- FDR:
-
False discovery rate
- eQTL:
-
Expression quantitative trait loci
- GO:
-
Gene ontology
- SMR:
-
Summary statistics-based Mendelian randomization
- MS:
-
Multiple sclerosis
- SLE:
-
Systemic lupus erythematosus
- T1D:
-
Type 1 diabetes
- CeD:
-
Celiac disease
- CD:
-
Crohn’s disease
- IBD:
-
Inflammatory bowel disease
- UC:
-
Ulcerative colitis
- PD:
-
Parkinson’s disease
- AD:
-
Alzheimer’s disease
References
Brown RH, Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162–72.
van Es MA, Hardiman O, Chio A, Al-Chalabi A, Pasterkamp RJ, Veldink JH, van den Berg LH. Amyotrophic lateral sclerosis. Lancet. 2017;390(10107):2084–98.
Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7(9):710–23.
Jaiswal MK. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med Res Rev. 2019;39(2):733–48.
Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019;18(2):211–20.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science (New York, NY). 2016;353(6301):777–83.
Thonhoff JR, Simpson EP, Appel SH. Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr Opin Neurol. 2018;31(5):635–9.
Zhao W, Beers DR, Appel SH. Immune-mediated mechanisms in the pathoprogression of amyotrophic lateral sclerosis. J Neuroimmune Pharmacol. 2013;8(4):888–99.
Zhao W, Beers DR, Hooten KG, Sieglaff DH, Zhang A, Kalyana-Sundaram S, Traini CM, Halsey WS, Hughes AM, Sathe GM, et al. Characterization of gene expression phenotype in amyotrophic lateral sclerosis monocytes. JAMA Neurol. 2017;74(6):677–85.
Pagani MR, Gonzalez LE, Uchitel OD. Autoimmunity in amyotrophic lateral sclerosis: past and present. Neurol Res Int. 2011;2011:497080.
Turner MR, Goldacre R, Ramagopalan S, Talbot K, Goldacre MJ. Autoimmune disease preceding amyotrophic lateral sclerosis: an epidemiologic study. Neurology. 2013;81(14):1222–5.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, et al. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016;8(347):347ra393.
Fredi M, Cavazzana I, Biasiotto G, Filosto M, Padovani A, Monti E, Tincani A, Franceschini F, Zanella I. C9orf72 intermediate alleles in patients with amyotrophic lateral sclerosis, systemic lupus erythematosus, and rheumatoid arthritis. NeuroMolecular Med. 2019;21(2):150–9.
Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, Rujescu D, Werge T, van de Bunt M, Morris AP, et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92(2):197–209.
Yokoyama JS, Wang Y, Schork AJ, Thompson WK, Karch CM, Cruchaga C, McEvoy LK, Witoelar A, Chen CH, Holland D, et al. Association between genetic traits for immune-mediated diseases and Alzheimer disease. JAMA Neurol. 2016;73(6):691–7.
Witoelar A, Jansen IE, Wang Y, Desikan RS, Gibbs JR, Blauwendraat C, Thompson WK, Hernandez DG, Djurovic S, Schork AJ, et al. Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol. 2017;74(7):780–92.
Nicolas A, Kenna KP, Renton AE, Ticozzi N, Faghri F, Chia R, Dominov JA, Kenna BJ, Nalls MA, Keagle P, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron. 2018;97(6):1268–83 e1266.
Shrine N, Portelli MA, John C, Soler Artigas M, Bennett N, Hall R, Lewis J, Henry AP, Billington CK, Ahmad A, et al. Moderate-to-severe asthma in individuals of European ancestry: a genome-wide association study. Lancet Respir Med. 2019;7(1):20–34.
Patsopoulos NA, Baranzini SE, Santaniello A, Shoostari P, Cotsapas C, Wong G, Beecham AH, James T, Replogle J, Vlachos IS. et al. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science (New York, NY). 2019;365(6460):eaav7188.
Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, Capon F, Ding J, Li Y, Tejasvi T, Gudjonsson JE, et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet. 2012;44(12):1341–8.
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376–81.
Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, Martín J, Fairfax BP, Knight JC, Chen L, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet. 2015;47(12):1457–64.
Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE, Mentch FD, Qiu H, Glessner JT, Thomas KA, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011;7(9):e1002293.
Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, Heap GA, Adány R, Aromaa A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42(4):295–302.
de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji SG, et al. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat Genet. 2017;49(2):256–61.
Benyamin B, He J, Zhao Q, Gratten J, Garton F, Leo PJ, Liu Z, Mangelsdorf M, Al-Chalabi A, Anderson L, et al. Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nat Commun. 2017;8(1):611.
Lu Q, Li B, Ou D, Erlendsdottir M, Powles RL, Jiang T, Hu Y, Chang D, Jin C, Dai W, et al. A powerful approach to estimating annotation-stratified genetic covariance via GWAS summary statistics. Am J Hum Genet. 2017;101(6):939–64.
Shi H, Mancuso N, Spendlove S, Pasaniuc B. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am J Hum Genet. 2017;101(5):737–51.
Yang J, Weedon MN, Purcell S, Lettre G, Estrada K, Willer CJ, Smith AV, Ingelsson E, O’Connell JR, Mangino M, et al. Genomic inflation factors under polygenic inheritance. Eur J Human Genetics. 2011;19(7):807–12.
Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55(4):997–1004.
Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, Kelsoe JR, O’Donovan MC, Furberg H, Schork NJ, et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9(4):e1003449.
Andreassen OA, Thompson WK, Schork AJ, Ripke S, Mattingsdal M, Kelsoe JR, Kendler KS, O’Donovan MC, Rujescu D, Werge T, et al. Improved detection of common variants associated with schizophrenia and bipolar disorder using pleiotropy-informed conditional false discovery rate. PLoS Genet. 2013;9(4):e1003455.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300.
Efron B. Size, power and false discovery rates. Ann Stat. 2007;35(4):1351–77.
Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, De T, Coin L, de Silva R, Cookson MR, et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014;17(10):1418–28.
Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science (New York, NY). 2015;348(6235):648–60.
Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39(Database issue):D712–7.
Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue A, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–102.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, DeStafano AL, Bis JC, Beecham GW, Grenier-Boley B, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8.
Stephenson J, Nutma E, van der Valk P, Amor S. Inflammation in CNS neurodegenerative diseases. Immunology. 2018;154(2):204–19.
Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol (Baltimore, Md : 1950). 2011;186(8):4973–83.
Gadoth A, Nefussy B, Bleiberg M, Klein T, Artman I, Drory VE. Transglutaminase 6 antibodies in the serum of patients with amyotrophic lateral sclerosis. JAMA Neurol. 2015;72(6):676–81.
Jackson JR, Eaton WW, Cascella NG, Fasano A, Kelly DL. Neurologic and psychiatric manifestations of celiac disease and gluten sensitivity. Psychiatric Quarterly. 2012;83(1):91–102.
Visser AE, Pazoki R, Pulit SL, van Rheenen W, Raaphorst J, van der Kooi AJ, Ricaño-Ponce I, Wijmenga C, Otten HG, Veldink JH, et al. No association between gluten sensitivity and amyotrophic lateral sclerosis. J Neurol. 2017;264(4):694–700.
Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience. 2007;144(3):991–1003.
Tanaka H, Shimazawa M, Takata M, Kaneko H, Tsuruma K, Ikeda T, Warita H, Aoki M, Yamada M, Takahashi H, et al. ITIH4 and Gpx3 are potential biomarkers for amyotrophic lateral sclerosis. J Neurol. 2013;260(7):1782–97.
Piątek-Guziewicz A, Zagrodzki P, Paśko P, Krośniak M, Ptak-Belowska A, Przybylska-Feluś M, Mach T, Zwolińska-Wcisło M. Alterations in serum levels of selected markers of oxidative imbalance in adult celiac patients with extraintestinal manifestations: a pilot study. Polish Archives Internal Med. 2017;127(7–8):532–9.
Pascual V, Dieli-Crimi R, López-Palacios N, Bodas A, Medrano LM, Núñez C. Inflammatory bowel disease and celiac disease: overlaps and differences. World J Gastroenterol. 2014;20(17):4846–56.
Philpott DJ, Sorbara MT, Robertson SJ, Croitoru K, Girardin SE. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14(1):9–23.
Fritz T, Niederreiter L, Adolph T, Blumberg RS, Kaser A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut. 2011;60(11):1580–8.
Sheykhansari S, Kozielski K, Bill J, Sitti M, Gemmati D, Zamboni P, Singh AV. Redox metals homeostasis in multiple sclerosis and amyotrophic lateral sclerosis: a review. Cell Death Dis. 2018;9(3):348.
Yiangou Y, Facer P, Durrenberger P, Chessell IP, Naylor A, Bountra C, Banati RR, Anand P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006;6:12.
Mariosa D, Kamel F, Bellocco R, Ye W, Fang F. Association between diabetes and amyotrophic lateral sclerosis in Sweden. Eur J Neurol. 2015;22(11):1436–42.
Kioumourtzoglou MA, Rotem RS, Seals RM, Gredal O, Hansen J, Weisskopf MG. Diabetes mellitus, obesity, and diagnosis of amyotrophic lateral sclerosis: a population-based study. JAMA Neurol. 2015;72(8):905–11.
Zeng P, Wang T, Zheng J, Zhou X. Causal association of type 2 diabetes with amyotrophic lateral sclerosis: new evidence from Mendelian randomization using GWAS summary statistics. BMC Med. 2019;17(1):225.
Lekoubou A, Matsha TE, Sobngwi E, Kengne AP. Effects of diabetes mellitus on amyotrophic lateral sclerosis: a systematic review. BMC Research Notes. 2014;7:171.
Miller ZA, Sturm VE, Camsari GB, Karydas A, Yokoyama JS, Grinberg LT, Boxer AL, Rosen HJ, Rankin KP, Gorno-Tempini ML, et al. Increased prevalence of autoimmune disease within C9 and FTD/MND cohorts: completing the picture. Neurology(R) Neuroimmunol Neuroinflammation. 2016;3(6):e301.
Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, Julier C, Morahan G, Nerup J, Nierras C, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet. 2009;41(6):703–7.
Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet. 2015;47(9):979–86.
Trynka G, Hunt KA, Bockett NA, Romanos J, Mistry V, Szperl A, Bakker SF, Bardella MT, Bhaw-Rosun L, Castillejo G, et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat Genet. 2011;43(12):1193–201.
Daoud H, Belzil V, Martins S, Sabbagh M, Provencher P, Lacomblez L, Meininger V, Camu W, Dupré N, Dion PA, et al. Association of long ATXN2 CAG repeat sizes with increased risk of amyotrophic lateral sclerosis. Arch Neurol. 2011;68(6):739–42.
Sproviero W, Shatunov A, Stahl D, Shoai M, van Rheenen W, Jones AR, Al-Sarraj S, Andersen PM, Bonini NM, Conforti FL, et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging. 2017;51(178):e171–8 e179.
Snook J. Are the inflammatory bowel diseases autoimmune disorders? Gut. 1990;31(9):961–3.
Sirabella R, Valsecchi V, Anzilotti S, Cuomo O, Vinciguerra A, Cepparulo P, Brancaccio P, Guida N, Blondeau N, Canzoniero LMT, et al. Ionic homeostasis maintenance in ALS: focus on new therapeutic targets. Front Neurosci. 2018;12:510.
Kuo JJ, Siddique T, Fu R, Heckman CJ. Increased persistent Na(+) current and its effect on excitability in motoneurones cultured from mutant SOD1 mice. J Physiol. 2005;563(Pt 3):843–54.
Tamura N, Kuwabara S, Misawa S, Kanai K, Nakata M, Sawai S, Hattori T. Increased nodal persistent Na+ currents in human neuropathy and motor neuron disease estimated by latent addition. Clin Neurophysiol. 2006;117(11):2451–8.
Zeng L, Zhang D, McLoughlin HS, Zalon AJ, Aravind L, Paulson HL. Loss of the spinocerebellar ataxia type 3 disease protein ATXN3 alters transcription of multiple signal transduction pathways. PLoS One. 2018;13(9):e0204438.
Du Y, Wen Y, Guo X, Hao J, Wang W, He A, Fan Q, Li P, Liu L, Liang X, et al. A genome-wide expression association analysis identifies genes and pathways associated with amyotrophic lateral sclerosis. Cell Mol Neurobiol. 2018;38(3):635–9.
Lan ZJ, Hu Y, Zhang S, Li X, Zhou H, Ding J, Klinge CM, Radde BN, Cooney AJ, Zhang J, et al. GGNBP2 acts as a tumor suppressor by inhibiting estrogen receptor α activity in breast cancer cells. Breast Cancer Res Treat. 2016;158(2):263–76.
Gibson SB, Abbott D, Farnham JM, Thai KK, McLean H, Figueroa KP, Bromberg MB, Pulst SM, Cannon-Albright L. Population-based risks for cancer in patients with ALS. Neurology. 2016;87(3):289–94.
Wang D, Chan CC, Cherry S, Hiesinger PR. Membrane trafficking in neuronal maintenance and degeneration. Cellular Molecular Life Sci. 2013;70(16):2919–34.
Kiral FR, Kohrs FE, Jin EJ, Hiesinger PR. Rab GTPases and membrane trafficking in neurodegeneration. Current Biol. 2018;28(8):R471–r486.
Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AGL, Varela MA, Sathyaprakash C, Scaber J, Barbagallo P, et al. C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain. 2017;140(4):887–97.
Sundaramoorthy V, Sultana JM, Atkin JD. Golgi fragmentation in amyotrophic lateral sclerosis, an overview of possible triggers and consequences. Front Neurosci. 2015;9:400.
Atkin JD, Farg MA, Soo KY, Walker AK, Halloran M, Turner BJ, Nagley P, Horne MK. Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J Neurochem. 2014;129(1):190–204.
Ni J, Wu GD, Albenberg L, Tomov VT. Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol. 2017;14(10):573–84.
Erny D, Prinz M. Microbiology: gut microbes augment neurodegeneration. Nature. 2017;544(7650):304–5.
Ambrosini YM, Borcherding D, Kanthasamy A, Kim HJ, Willette AA, Jergens A, Allenspach K, Mochel JP. The gut-brain axis in neurodegenerative diseases and relevance of the canine model: a review. Front Aging Neurosci. 2019;11:130.
Acknowledgements
We thank the investigators who made the GWAS summary data publicly available and the participants who contributed to those studies.
Funding
This research was supported by the funding of the National Natural Science Foundation of China (Grant No. 81901294). The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript.
Author information
Authors and Affiliations
Contributions
ChunYu Li: design of the work, statistical analysis, interpretation of data, and drafting of the manuscript; TianMi Yang: analysis and revising of the manuscript; RuWei Ou: revising of the manuscript; QianQian Wei: revising of the manuscript; HuiFang Shang: design, conception, and review. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Summary data from all GWAS used in current study. Table S2. Genetic correlation between AD, ALS, PD and autoimmune disorders. Table S3. Risk loci associated with ALS conditional on autoimmune diseases. Table S4. eQTL revealing functional effects of shared risk SNPs in tissues from GTEx. Table S5. Functional annotation of shared risk genes. Figure S1. Manhattan-style plots showing the estimates of local genetic correlation between ALS and autoimmune disorders. Figure S2. Conditional Manhattan plots showing risk loci for ALS conditional on each autoimmune disease. Figure S3. Regional association plots for shared risk loci between ALS and each autoimmune disease
Additional file 2.
Supplementary Methods.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, C.Y., Yang, T.M., Ou, R.W. et al. Genome-wide genetic links between amyotrophic lateral sclerosis and autoimmune diseases. BMC Med 19, 27 (2021). https://doi.org/10.1186/s12916-021-01903-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12916-021-01903-y