
Abstract
Background
As an endemic shrub of the Qinghai-Tibetan Plateau (QTP), the distribution of Hippophae tibetana Schlecht. ranges between 2800 and 5200 m above sea level. As the most basal branch of the Hippophae genus, H. tibetana has an extensive evolutionary history. The H. tibetana is a valuable tree for studying the ecological evolution of species under extreme conditions.
Results
Here, we generated a high-quality chromosome-level genome of H. tibetana. The total size of the assembly genome is 917 Mb. The phylogenomic analysis of 1064 single-copy genes showed a divergence between 3.4 and 12.8 Mya for H. tibetana. Multiple gene families associated with DNA repair and disease resistance were significantly expanded in H. tibetana. We also identified many genes related to DNA repair with signs of positive selection. These results showed expansion and positive selection likely play important roles in H. tibetana’s adaptation to comprehensive extreme environments in the QTP. A comprehensive genomic and transcriptomic analysis identified 49 genes involved in the flavonoid biosynthesis pathway in H. tibetana. We generated transgenic sea buckthorn hairy root producing high levels of flavonoid.
Conclusions
Taken together, this H. tibetana high-quality genome provides insights into the plant adaptation mechanisms of plant under extreme environments and lay foundation for the functional genomic research and molecular breeding of H. tibetana.
Similar content being viewed by others


Background
Known as the Himalaya Plateau, the Qinghai-Tibet Plateau (QTP) is the world’s largest and highest plateau [1]. In the QTP, considerable geological uplifts have occurred seven times since the Pliocene [2]. For example, it has been estimated that the QTP mountains were uplifted three times between 0.6 and 1.3 million years ago (Mya) [3]. Increasing altitude has resulted in extensive glaciation on the QTP. Naynayxungla Glaciation, the largest glaciation in the QTP, began approximately 1.2 Mya and peaked between 0.6 and 0.8 Mya. The uplift of the Qinghai-Tibet Plateau (QTP) has led to significant climatic and environmental transformations in the plateau region. Low temperature, low oxygen, reduced pathogen incidence, and strong UV radiation now characterize the QTP conditions, creating an ideal environment for studying adaptive evolution [4, 5]. The diversifications of many endemic species are highly consistent with environmental changes and climate change [6]. A majority of genome-wide studies on adaptive evolution have been conducted on vertebrates and humans [7,8,9]. According to these studies, genes involved in hypoxia responses, energy metabolism, and skeletal development are undergoing positive selection and rapid evolution. Recently, a few studies have been conducted on wild plants in this region using whole-genome analysis to study adaptive evolution, including Tibetan hulless barley [10], wild barley [11], maca [12], and Eutrema [13].
As an original species on the QTP, H. tibetana is widely distributed in the Himalayas region [14]. In the Elaeagnaceae family, Hippophae is a small genus composed of five to seven species. Nitrogen-fixing is a function of all species of this genus [15]. The species of H. tibetana, which is morphologically uniform, lives in a variety of habitats ranging from 2800 to 5000 m in altitude [2]. Hippophae originated from ancient Mediterranean coast (the present Qinghai Tibet Plateau and Xinjiang) at 2500–4000 Mya. Phylogenetic analysis revealed that the divergence of H. tibetana from the other species of Hippophae genus occurred at around 3.15 million years ago [16]. The rapid rise of QTP may have altered the genetic factors of H. tibetana, reflecting their adaptation to the extreme environment.
Flavonoids from sea buckthorn (H. rhamnoides L.) were used to prevent and manage chronic diseases such as diabetes, cardiovascular disease, and cancer [17]. Sea buckthorn leaves, roots, stems, and fruits have high total concentration [18]. As a subgroup of flavonoids, flavonols are the most abundant in sea buckthorn. In sea buckthorn leaves and berries, the isorhamnetin glycosides are typically the most significant flavonols [19]. A variety of genetic and environmental factors influence flavonoid accumulation in sea buckthorn [18]. There are lots of genes involved in the flavonoid biosynthesis pathways, including phenylalanine ammonia lyase (PAL), cinnamic acid hydroxylase (C4H), 4-coumaric acid CoA ligase (4CL), chalcone synthase (CHS), chalcone isomerase (CHI), flavone synthase I and II (FNSI and FNS II), isoflavone synthase (IFS), flavanone-3-hydroxylase (F3H), flavonol 3′-hydroxylase (F3′H), flavonol 3′5′-hy-droxylase (F3′5′H), flavonol synthase (FLS), and dihydroflavonol 4-reductase (DFR). There is little information about the genes involved in flavonoid biosynthesis caused by historical genome duplication and gene family expansions and contractions. Genome assembly with annotations at the chromosome level could help identify key factors in flavonoid biosynthesis.
The draft genome of H. rhamnoides subsp. mongolia was assembled [20]. However, it has high heterozygosity (0.65%) and high rate of sequence repeats (> 65%). There were over 3000 fragmentary scaffolds in the genome of H. rhamnoides subsp. mongolia, which could easily have led to inaccuracies in genomic studies. In general, high-quality reference genomes for sea buckthorn species would be important for functional genomic studies.
Here, long-read sequencing technology, PacBio HIFI technology, and HiC sequencing technology were used to assemble a high-quality genome of H. tibetana with its high levels of repeat elements (> 65%). Using this newly obtained genome of H. tibetana, we performed comparative and evolutionary genomic analysis to clarify its origins and investigate evolutionary signals. In addition, by combining genomic and transcriptomic analysis, we identified key genes associated with the biosynthetic pathways of flavonoids in H. tibetana. We also confirmed the role of two genes (HtC4H1 and HtC4H2) involved in the flavonoid biosynthetic pathway. The high-quality genome of H. tibetana helped us better understand how H. tibetana has adapted to the complex conditions on the QTP, and our results laid foundation resources for the molecular breeding of H. tibetana medicinal materials with higher flavonoid content.
Results
Chromosome-level reference genome of H. tibetana
Using the PacBio Sequel II platform, we generated ~ 50 × long reads to obtain a high-quality genome for H. tibetana (2n = 2x = 24) (Fig. 1). The estimated genome size was 997.42 Mb based on Illumina short-read sequencing data (Table 1, Additional file 1: Fig. S1). Assembling the dataset with Hifiasm yielded a 957.16-Mb assembly, which is 95.9% the estimated genome size. There were 90 contigs generated from the assembly, giving the genome assembly a contig N50 of 36.40 Mb. Then, the PacBio sequence assembly was polished with 52.08 Gb (~ 52 ×) of Illumina sequencing reads to remove sequencing errors found in single-molecule sequence datasets. After that, a total of 100.78 Gb of Hi-C data were generated, which was integrated into the assembly (Fig. 2a). Consequently, 12 pseudochromosomes (scaffold N50 size, 88.98 Mb) of H. tibetana genome were successfully assembled, which contains nearly 92.99% (890.09 Mb) of the assembled sequences (Fig. 2b, Additional file 2: Table S1). The assembled sequences showed low GC content of 29.8%. Compared with the genome sequences of H. rhamnoides subsp. mongolia, our assembly (contigs N50 of 36.40 Mb) was obviously improved, which contained 30 scaffolds. Genome assembly coverage was evaluated by mapping high-coverage Illumina sequencing reads against the assembled genome. There was a 99.91% mapping rate for Illumina DNA reads and 97.03% for Illumina RNA reads.

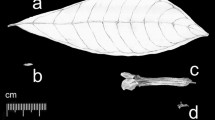
Sampling location and morphology of H. tibetana. a Sampling location of H. tibetana. b Photographs of H. tibetana trees. c Photographs of H. tibetana fruits

Overview of the H. tibetana genome. a Genome-wide all-by-all interactions among all sea buckthorn chromosomes. b (A) Gene density and distribution (non-overlapping window size, 100 kb); (B–E) gene expression levels (Log2 FPKM) in sea buckhorn stem, root, leaves, fruit; (F) density of repeats (non-overlapping window size, 100 kb); (G) density of Copia-type transposons (non-overlapping window size, 100 kb); (H) density of Gypsy-type transposons (non-overlapping window size, 100 kb); (I) sea buckthorn pseudo-chromosomes. c Insertion time distribution of LTR retrotransposons
The assembled H. tibetana genome was 211 Mb larger than the reported genome of H. rhamnoides subsp. sinensis. In the H. tibetana genome, transposable elements (TEs) were more abundant than in the H. rhamnoides subsp. sinensis genome. It was found that TEs accounted for 605 Mb, or 66.01%, of the assembled H. tibetana genome, as compared to 335 Mb of TEs in the genome of H. rhamnoides subsp. sinensis (47.48%).
Gene prediction and functional annotation
In H. tibetana, 31,340 protein-coding genes were annotated based on de novo, homologous, and RNA sequencing predictions. The average length of genes and coding sequences were 4387 bp and 1176 bp, respectively (Additional file 2: Table S2). According to mapping results, 96.8% (30,345) of the predicted genes have a corresponding function (Additional file 1: Fig. S2). In addition, the sea buckthorn genome was predicted to contain 391 miRNAs, 941 tRNAs, 3459 rRNAs, and 4425 snRNAs (Additional file 1: Fig. S3, Additional file 2: Table S3). We assessed the quality and completeness of this assembled genome using two strategies. First, the H. tibetana genome contained 237 (95.56%) core eukaryotic genes by a core eukaryotic gene mapping approach (CEGMA) analysis. Then, a 98.2% (1614 genes) BUSCO recovery score demonstrated the high completeness of the assembly of the H. tibetana genome (Additional file 2: Table S4). These parameters of the H. tibetana genome are better than other published Elaeagnaceae Juss. genomes.
Through multiple de novo prediction procedures, we identified and classified repeat sequences within the repeat library using RepeatMasker (Additional file 2: Table S5). Approximately, 65.77% of the H. tibetana sequences were identified as repetitive elements, including retrotransposons (51.11%), DNA transposons (3.3%), and unclassified elements (14.76%). The most common type of retrotransposons (50.48%) were long-terminal repeats (LTRs), of which Copia and Gypsy constituted 17.55% and 17.26% of the genome, respectively. In H. tibetana, retrotransposons might be associated with genome expansion. Based on H. tibetana genome sequences, we estimated the insertion dates of LTR retrotransposons to explore its evolutionary dynamics. In H. tibetana, LTR retrotransposons are most abundant at ~ 1.0 Mya (Fig. 2c). As a result of LTR retrotransposons’ proliferation, H. tibetana has probably evolved to become more diverse, speciated, and adapted to high altitude conditions.
Evolution and gene family expansion/contraction analysis
The genome sequences of H. tibetana and another 10 plants (H. rhamnoides subsp. mongolia, H. rhamnoides subsp. sinensis, Populus trichocarpa, Ziziphus jujube, Vitis vinifera, Fragaria vesca, Arabidopsis thaliana, Malus domestica, Carica papaya, and Citrus sinensis) were analyzed to determine the expansion and contraction of genes in the genome of H. tibetana. We totally identified 25,729 orthogroups from 11 plant genomes, 8602 of which were common between all 11 plants (Fig. 3a, b). H. tibetana genome contained 281 species-specific orthogroups, containing 857 genes. These 857 genes were found to be significantly enriched in a number of biological processes, including protein processing in the endoplasmic reticulum, plant-pathogen interaction, ribosome biogenesis in eukaryotes, and ubiquitin-mediated proteolysis (Additional file 1: Fig. S3).

Characterization of H. tibetana genome evolution and gene family expansion. a Distribution of gene numbers and family sizes in H. tibetana and 10 other representative species. b Venn diagram showing unique and shared gene families between genomes of H. tibetana and 4 close relatives. c A species tree on the basis of 1064 single-copy orthologues from H. tibetana and 10 other representative species. The black numbers represent the divergence times from present. Green and red indicate the numbers of significantly (p < 0.05) expanded and contracted gene families. d KEGG functional classification of contracted gene families. e KEGG functional classification of expanded gene families
The 1064 orthogroups with single-copy genes were identified from the 8602 common orthogroups. The single copy genes of the 11 plants were used to construct a phylogenetic tree (Fig. 3c). It is obvious that all 3 species of Hippophae L. (H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis) are clustered together. Also, in comparison with H. tibetana, H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis show the closest distance. According to the divergent analysis, H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis diverged from Ziziphus jujube at 79.7 Mya. The phylogenetic evolutionary relationship revealed that the H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis diverged at 7.6 Mya.
As a result of further analysis of gene clusters, numerous genes were found to have expanded or contracted in the 11 genomes. For the H. tibetana, a total of 430 expanded and 1426 contracted orthogroups were identified, respectively (Additional file 2: Table S6). Among 430 expanded orthogroups, 201 orthogroups with 1298 genes were significantly expanded (p < 0.01). According to the functional analysis of these 1298 genes (Fig. 3d), protein processing in the endoplasmic reticulum was the most enriched biological process. Among these expanded genes, a tryptophan decarboxylase (TDC) gene was found, which is involved in melatonin biosynthesis. Five CBL-interacting protein kinase (CIPK) genes were found, which play important roles in abiotic stress response. Two damage tolerance protein genes (rad31-1 and rad31-2) were UBA-related genes required for DNA damage tolerance. Five genes involved in flavonoid biosynthesis were found. Three calcium-binding protein CML10 genes regulate cold tolerance. We identified a gene (Chr08.46) that is involved in photooxidative stress responses and cell death induced by UV-C. In addition, we also discovered genes associated with disease resistance in plants, including 4 pathogenesis-related gene 1 and 2 fatty acid 2-hydroxylase 1-like (FAH2, Chr09.2289, Chr11.212) genes, produce reactive oxygen species after chitin treatment. A total of 6 genes encoding alpha-glucan endo-1,3-beta-glucosidase have been found, which is considered as a defense factor against fungal pathogens. Five dirigent protein-like (DIR) genes were identified, which are involved in the plant disease-resistance response.
There is often an association between the adaptive divergence of closely related species and the contraction in the size of particular gene families. A total of 1426 gene families were identified as significantly contracted genes in the H. tibetana genome compared to the genomes of 10 species (p < 0.05) (Additional file 2: Table S6). Based on KEGG enrichment analysis results, contracted genes were significantly enriched in plant secondary metabolite biosynthesis, cyanoamino acid metabolism, and starch and sucrose metabolism (Fig. 3e). Among these contracted genes, 104 genes were annotated as resistance-related genes, including 8 NBS-LRR genes, 13 LRR-RLK genes, 45 RLK genes, 2 RLP genes, 6 GLR genes, 13 GsSRK genes, 7 CRK genes, 3 GDSL esterase/lipase genes, and 7 laccase genes.
A whole-genome duplication (WGD) event is a key factor in the evolution of plants, animals, fungi, and other organisms. In order to investigate WGD in H. tibetana, we selected four species to perform a comparative genomics analysis. Based on the distributions of Ks of paralogous gene pairs in the H. tibetana genome, two recent clear peaks were found at 0.28 and 0.4 (Fig. 4a). There were two similar Ks peaks in the genomes of two closely related species, E. mollis and H. rhamnoides. Therefore, these two peaks might represent two closely WGD events in H. tibetana. These two WGD events were estimated to have occurred ~ 26 Mya and ~ 38 Mya. Phylogenetic evolutionary analysis indicated that these two WGD events occurred after Hippophae L. diverged from Jujuba.

Identification of the WGDs in H. tibetana genome. a Ks distribution from orthologs between H. tibetana and other three species. b Gene Ontology (GO) functional classification of genes with WGDs. c KEGG functional classification of genes with WGDs
As a driving force for plant evolution, WGD has the potential to endow genes with new functions. GO enrichment analysis of genes associated with WGD events showed that these genes were significantly enriched into protein catabolic process, cellular protein catabolic process, and proteolysis involved in cellular protein catabolic process (Fig. 4b, c). Our KEGG enrichment analysis found that genes associated with WGD significantly enriched into plant hormone signal transduction, MAPK signaling pathways, fatty acid biosynthesis, and flavonoid biosynthesis (F3′H, CHS, C4H, CHI, DFR) (Fig. 4c). Thus, WGD events may related with high level of fatty acid and flavonoid content in sea buckthorn.
Candidate genes associated with high-altitude adaptation of the H. tibetana
Genes under positive selections were commonly associated with abiotic stresses, such as low temperature, low oxygen, reduced pathogen incidence, and strong UV radiation. The adaptive divergence of orthologs that show signs of positive selection usually occurs as a result of positive selection. Our study employed genomic sequences from H. tibetana and its 5 close species to conduct a positive selection analysis. We identified genes that show signs of positive selection from 1064 single-copy orthologs using the PAML 4 package’s branch-site model. The H. tibetana genome contained 212 genes possibly under positive selection (ω > 1, p < 0.05) (Additional file 2: Table S7). According to KEGG functional annotation, several of these genes are related to homologous recombination (RAD3-like DNA-binding helicase protein, DRD1 DEFECTIVE IN MERISTEM SILENCING 1, XRCC2 homolog of X-ray repair cross complementing 2, POLD3 DNA-directed DNA polymerase, RecQl3 DEAD/DEAH box RNA helicase family protein), Mismatch repair (5′-3′ exonuclease family protein, POLD3 DNA-directed DNA polymerase), ubiquinone and other terpenoid-quinone biosyntheses, base excision repair (poly ADP-ribose polymerase 3, POLD3 DNA-directed DNA polymerase), and plant-pathogen interaction (MKK6 MAP kinase kinase 6, calcium-binding EF-hand family protein). We identified genes that encode the RAD3-like DNA-binding helicase protein, DNA-directed DNA polymerase (POLD3), homolog of X-ray repair cross complementing 2 (XRCC2), DEAD/DEAH box RNA helicase family protein, 5′-3′ exonuclease family protein, and poly ADP-ribose polymerase 3, providing evidence that adaptive evolution in plants depends on positive selection of genes. In addition, two plant-pathogen interaction genes (MKK6 MAP kinase kinase 6 and calcium-binding EF-hand family protein) were also under positive selection, indicating that positive selection of genes may have a close correlation with plant-pathogen interaction in TQP.
Genome comparison between H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis
A variety of genetic variations, such as insertions and deletions, cause genetic diversity and phenotypic variation. Compared to H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis, H. tibetana exhibited obvious phenotypic differences, which allowed us to analyze the genomic variations between these three species. Full-genome alignment results revealed 237 syntenic blocks of H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis. These syntenic blocks contained 21,623 and 21,905 conserved genes covering 68.99% and 69.89% of the H. tibetana genome, respectively (Fig. 5a). In H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis, there are many collinear blocks, suggesting there is a high level of gene collinearity between the genomes (Fig. 5a). We identified 9,932,195 SNPs and 11,120,000 SNPs in H. tibetana genome compared with H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis genome (Fig. 5b, c). In addition, we identified 58,645 inversions, 13,967 deletion, and 11,305 insertion in H. tibetana genome compared with H. rhamnoides subsp. mongolia genome (Fig. 5b). Less deletion and insertion were found between H. tibetana and H. rhamnoides subsp. sinensis genome (Fig. 5b). Following this, we conducted structural variation analysis in the exon, promoter (2 kb up-5′UTR) and downstream (1 kb down-3′UTR) regions of gene in the H. tibetana genome. Compared with H. rhamnoides subsp. mongolia genome, we identified 2900 SV in the exon regions, 11,146 SV in the promoter regions, and 8616 SV in the downstream regions (Fig. 5b). Less SVs were found between H. tibetana and H. rhamnoides subsp. sinensis genome (Fig. 5c, d). H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis genomes show great variation possibly explaining their phenotypic differences.

The comparison of H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis genomes. a Syntenic blocks share between the H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis. b The numbers of SNPs, delection, insertion, and inversion in the H. tibetana genome compared with H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis. genome. c The distribution of structural variation (SV) in the exon, intron, UTR, promoter (2 kb up-5′UTR), and downstream (1 kb down-3′UTR) regions of genes in the H. tibetana genome compared with H. rhamnoides subsp. mongolia genome. d The distribution of structural variation (SV) in the exon, intron, promoter (2 kb up-5′UTR), and downstream (1 kb down-3′UTR) regions of genes in the H. tibetana genome compared with H. rhamnoides subsp. sinensis genome
Investigation of genes involved in the flavonoid biosynthetic pathway
Past researches reported the contents of phenylpropanoids and flavonoids were associated with the tolerance to UV light of Tibetan peach, Arabidopsis, and rice in TPQ [6, 21]. In plants, flavonoid biosynthetic genes have been identified and characterized (Fig. 6a). In the genome of H. tibetana, 49 homologs of genes associated with flavonoid biosynthesis were identified (Additional file 2: Table S8). Gene cluster analysis results showed that all these genes were divided into 3 groups (Fig. 6b). Most genes (44 genes) were expressed in all 4 different tissues, according to gene expression analysis. The root had higher expression levels for 17 genes involved in flavonoid biosynthesis than other tissues (Fig. 6b). In the fruit, 13 genes were more highly expressed in the flavonoid biosynthesis pathway (Fig. 6b). For example, the fruit exhibited extremely high expression levels of a F3H gene (HtChr08.706) (FPKM > 2571). In contrast, 2 C4H (HtChr04.654 and HtChr07.1953) genes have higher expression levels in the roots, leaves, and stem tissues, indicating the 2 genes might be related with the more flavonoid content in the roots, leaves, and stem of H. tibetana (Fig. 6c, d).

Genes associated with flavonoid biosynthesis in sea buckthorn. a Identification of structural flavonoid biosynthesis genes in H. tibetana. b Expression level of genes involved in the flavonoid biosynthesis pathway was detected in four different tissues of H. tibetana. The red colors indicated high expression levels. The blue colors indicated low expression levels. c the total flavonoid content of four different tissues of H. tibetana. d Maximum-likelihood trees of C4H family genes that were constructed using the amino acid sequences with 1000 bootstrap repeats. e Induction process of sea buckthorn hairy roots. CK, empty vector; OE-HtC4H1, overexpression lines (K599-1302-HtC4H1); OE-HtC4H2, overexpression lines (K599-1302-HtC4H2). f Relative expression levels of HtC4H1 and HtC4H2 in normal and transgenic hairy roots determined by qPCR. **p < 0.01, *p < 0.05. g Determination of flavonoids in CK and OE of hairy roots
Genomics and RNA sequencing provide valuable information for metabolic engineering that can enhance flavonoid content (Additional file 1: Figs. S4 and S5). The phenolic biosynthesis pathway starts with C4H, which is important for flavonoid accumulation in many plants. The flavonoid content of H. tibetana hairy roots was increased by overexpressing two C4H genes (HtC4H1 and HtC4H2) (Fig. 6e–g). We found that transgenic hairy roots showed higher flavonoid content compared with normal hairy roots (Fig. 6g). Hence, sea buckthorn flavonoids can be produced by cultivating hairy roots on a large scale.
Discussion
Genomics sequences and annotations are fundamental to many genetic studies, including examining the inheritance of traits, comparing genetics, and studying evolution [22, 23]. Using PacBio, Hi-C, and Illumina data, we have assembled a high-quality chromosomal-level genome of H. tibetana, which is highly complete in protein-coding genes, repetitive sequences, and contigs. As a result, the genome sequences are excellent resources for studying a wide range of genetic studies in the Hippophae species and its related species. A larger genome size is found in H. tibetana than in other Hippophae species [20, 24]. Among Hippophae species, H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis experienced two very recent WGD events, which occurred at ~ 26 Mya and ~ 38 Mya. In close species, Elaeagnus moorcroftii and Elaeagnus mollis also have experienced two very recent WGD events [25]. All these results indicated that these two WGD events were species-special and may play important roles in Elaeagnaceae.
Past research revealed that significant expansion of LTR retrotransposons was found in the genome of several reported plants living in the Qinghai Tibet Plateau. For example, C. himalaica exhibits a dramatic increase in LTR retrotransposons following a dramatic elevational and climatic change during Miocene to Pliocene [26]. A higher proportion of retrotransposons were also found in the whole genome of Tibetan hulless barley [10], wild barley [11], maca [12], and Eutrema heterophyllum [13]. We also found that LTR retrotransposons have insertion at ~ 1.0 Mya in the H. tibetana genome, which is similar to these in H. rhamnoides subsp. mongolia [20]. The significant expansion of LTR retrotransposons may be related to the extreme environmental adaptability of plants in the QTP.
Diversification and adaptation of species are crucially influenced by gene family expansion and contraction. In Tibetan semi-wild wheat [27] and C. himalaica [26], genes involved in DNA repair and protein ubiquitination pathways were significantly expanded, including DNA excision repair protein ERCC-1, AP endonuclease 2 protein (APEX2), DNA polymerase delta subunit 2 (POLD2), DNA mismatch repair protein MLH1, and DNA repair and recombination protein RAD54. In the Eutrema heterophyllum genome [13], five expanded genes (NBS1, RPA32, SNI1, KU80, XRCC4) were found in the DNA repair pathway. In H. tibetana, five CBL-interacting protein kinase (CIPK) genes, two damage tolerance protein genes (rad31), three calcium-binding protein CML10 genes, and one phototropin-1 gene, which related with DNA repair, were also identified as expanded genes. In addition, a number of expanded genes were also found to be associated with disease resistance in plants, including four pathogenesis-related gene 1, two fatty acid 2-hydroxylase 1-like genes, six glucan endo-1,3-beta-glucosidase genes, and five dirigent protein-like genes. Overall, these results suggest that gene expansion is a key mechanism for plant adaptation to high altitudes. Otherwise, 8 NBS-LRR genes, 13 LRR-RLK genes, and 45 RLK genes were significantly contracted in H. tibetana. A similar observation was made about the genome of C. himalaica, where disease resistance R genes were also observed to show significant contraction [26]. In wheat, the Pm21 gene encodes a CC-NBS-LRR protein that confers resistance to powdery mildew [28]. The Arabidopsis LRR-RLK IOS1 plays a critical role in pattern-triggered immunity triggered by BAK1 [29]. For rice, Pi-d2 encodes a G-type RLK protein that provides resistance to Magnaporthe grisea [30]. Two RLP genes, which encode receptor-like proteins, are also involved in contracted genes. An important role played by RLP1.1 is in responding to Puccinia striiformis f. sp. Tritici [31]. In addition, six GLR-contracted genes were also found in H. tibetana, which have a role in innate immune response. Fov7, encoding a GLR protein, confers resistance to Fusarium oxysporum f. sp. vasinfectum in upland cotton [32]. These genes may have evolved rapidly as a means of adapting to the rapidly changing pathogen spectrum of different species in QTP.
The adaptive divergence of orthologous genes has been accompanied by positive selection [26]. In the present study, an analysis of the H. tibetana genome totally identified 212 genes potentially under positive selection. A KEGG pathway enrichment analysis showed that these genes were associated with DNA repair, mismatch repair, base excision repair, and plant-pathogen interaction in the H. tibetana genome, which is similar with these in previous studies of C. himalaica [26]. For example, CheY-like genes demonstrated positive selection during adaptive evolution in C. himalaica. QTP is exposed to extremely intense UV radiation that could negatively impact plant growth and development, causing DNA, RNA, and protein damage [26]. A total of 7 genes under positive selection in the H. tibetana genome are related with the DNA repair pathway and homologous recombination pathway. It has also been found that two of them encode the DNA-binding helicase protein RAD3 and the DNA-directed polymerase protein POLD3, which was a component of DNA repair [26]. Furthermore, animals and plants are vulnerable to high UV-B radiation on the QTP. In Tibetan chicken, Tibetan hot-spring snake, and Tibetan barley, DNA repair pathways played an important role in adapting to high altitudes [7]. All these results indicated an important mechanism of genes under positive selection for high-attitude adaptive of H. tibetana.
In the Hippophae genus, flavonoids are a major subgroup of bioactive compounds that may play a key role in the prevention and management of chronic diseases such as diabetes and cardiovascular disease [17]. Flavonoids were found in sea buckthorn berries at a level of 1680–8570 mg/kg, more than in other plants with high flavonoids [18]. In many species, the genes and enzymes involved in flavonoid biosynthesis have been studied. Our study first identified and characterized 49 genes involved in the flavonoid biosynthesis pathway based on the genomic and transcriptomic analysis. Additionally, the WGD event can contribute to the formation and evolution of plant-specific pathways for metabolite biosynthesis in tea, yew, and Scutellaria baicalensis [22, 23]. In H. tibetana, we found five genes related to flavonoid biosynthesis undergone WGD, including F3′H, CHS, C4H, CHI, and DFR. Previous studies revealed the role of F3′H, CHS, C4H, CHI, and DFR in flavonoid biosynthesis in many plants [21]. A WGD event provided resources for the flavonoid biosynthetic pathway in H. tibetana.
The genomic and transcriptomic datasets of H. tibetana provided new insights and valuable tools into the flavonoid biosynthetic pathway. Recently, there have been no studies about manipulating flavonoid biosynthesis in H. tibetana. Flavonoid biosynthesis involves multiple enzymes, suggesting that metabolic flux may be limited by more than one enzyme. The C4H gene enhances the synthesis of flavonoids in Nicotiana tabacum, Scutellaria baicalensis, Ginkgo, and tartary buckwheat [33,34,35,36]. In this study, we first obtained transgenic H. tibetana hairy roots with significantly increased flavonoid content through overexpressing multiple genes (HtC4H1 and HtC4H2) related to the flavonoid biosynthetic pathway. Consequently, the genes involved in flavonoid biosynthesis may be candidates for metabolically engineered flavonoid enhancement.
Conclusions
This study generated one high-quality reference genome of H. tibetana, with a contig N50 of 36.4 Mb. The high-quality chromosome-level genome provides a reference for the understanding of high-altitude adaptation. In addition, transcriptomic and genomic investigations also provided an in-depth insight into flavonoid biosynthesis. Genomic data of H. tibetana will provide essential resources for further functional study with genome editing and improving H. tibetana as an economic crop for edible food and traditional Chinese medicine.
Methods
Plant materials, genomic DNA extraction, and sequencing
The H. tibetana plant was obtained from hongyuan county (32° 26′ 2.17″ N, 102° 22′ 11.61″ E), Sichuan Province in Eastern China. Following the manufacturer’s instructions, we extracted the genomic DNA from the young leaves of H. tibetana plants using a MiniBEST Plant Genomic DNA Extraction Kit (TaKaRa). The extracted cDNA was detected by a NanoDrop™ One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA) and Qubit® 3.0 Fluorometer (Invitrogen, USA). After that, we used the BluePippin system (Sage Science, USA) to select and collect long DNA fragments. A 20-kb library was constructed according to the manufacturer’s instructions. This library was sequenced using the PacBio Sequel II system. We constructed Hi-C libraries according to the methods in past researches [37,38,39]. A formaldehyde solution was used to fix the leaf samples. Cross-linked DNA was digested with 400 U of DpnII restriction enzyme at 37 °C. T4 DNA ligase (NEB) was used for DNA ligation for DNA with biotin labeled. After reverse crosslinking, the DNA fragments were purified and fragmented to 300–500 bp. We used Dynabeads® M-280 Streptavidin (Life Technologies) to separate biotin-labeled DNA fragments. Hi-C libraries were constructed and sequenced on an Illumina HiSeq X Ten sequencer. According to the description in the past research, DNA libraries were constructed for Illumina sequencing [20]. Briefly, DNA segmentation, repairing DNA ends, and adding poly (A) adaptors were performed. Final DNA libraries were sequenced on the Illumina HiSeq 2500 system.
Genome assembly and quality assessment
Illumina short reads were used for evaluating the genome size, heterozygosity ratio, and repeat sequence ratio using k-mer distribution analysis (k = 17). The CCS software is used to process the sequencing data of the Pacbio CCS platform to generate high-precision HiFi data. Based on PacBio reads, we assembled the contig-level genome using Hifiasm (parameters “-l 2 -k 51 -w 51 --write-paf --write-ec --h1 --h2”) [40]. Hi-C sequencing data were mapped to contigs sequences using the hiccup software to evaluate the quality of HiC data [41]. The ALLHiC (v0.9.8) software was used to assemble the chromosome-level genome using Hi-C data [42]. Fine adjustment of orientation results with the JuiceBox software [43]. We assessed the completeness of the assembled chromosome-level genome using conserved plant genes from the BUSCO (v3.0.2) and CEGMA (v2.5) databases. In order to assess coverage rate and average depth, BWA (v0.7.8) was used to map Illumina short reads to the assembled genome [44].
Genome annotations
To annotate repetitive sequences in the assembled H. tibetana genome, we used RepeatModeler (v.1.0.5), RepeatScout (v.1.0.5), and LTR_FINDER (v.1.07) to build a de novo repeat library. Then, the repeat library (Repbase v15.02) was used to identify homologous repeat by RepeatMasker (v.4.0.7) and RepeatProteinMask (v.4.0.7) [45]. Tandem Repeats Finder (v4.09) was used to identify the tandem repeats in the H. tibetana genome [46].
We used 5 ab initio gene prediction programs for de novo prediction, including Augustus (v3.2.3), Geneid (v1.4), Genescan (v1.0), GlimmerHMM (v3.0.2), and SNAP (v2013.11.29). tBlastn (v2.2.26, E-value < 10e − 5) was used to predict gene models by aligning the protein sequences of five species (Arabidopsis thaliana, Elaeagnus angustifolia, Fragaria vesca, H. rhamnoides, Malus domestica) against the H. tibetana genome. RNA sequencing data from four different tissues were mapped to the H. tibetana genome for gene prediction using Tophat [47]. We conducted a comparative analysis of genes against the SwissProt, TrEMBL, and non-redundant protein databases using BLASTP, with a significance threshold set at an E-value of ≤ 1E − 05. The best hits were used to annotate the gene function. The GO and KEGG annotations for each gene were obtained by comparison with homologs in the GO and KEGG databases. The tRNAscan-SE (v1.4) and INFERNAL (v1.1.2) software were used to predict noncoding RNAs [48, 49].
Comparative genomics and genome evolution analysis
Protein sequences from the ten genomes (H. rhamnoides subsp. mongolia, H. rhamnoides subsp. sinensis, Populus trichocarpa, Ziziphus jujube, Vitis vinifera, Fragaria vesca, Arabidopsis thaliana, Malus domestica, Carica papaya, Citrus sinensis) were downloaded from Ensembl Plants (http://plants.ensembl.org/index.html). Orthologous gene families were obtained using OrthoMCL (v1.4) [50]. MUSCLE (v3.8.31) was used to obtain a super-alignment matrix by aligning single-copy gene families. Then, an ML phylogenetic tree was constructed using MEGA7 [51]. Based on the TimeTree database (http://www.timetree.org/), we used MCMCTree to calculate the divergence time. The CAFE (v4.2) software was used to detect expanded and contracted gene families [52]. H. tibetana genomes were searched with LTR_FINDER to identify intact LTR retrotransposons [53]. The insertion times of LTR retrotransposons were calculated using LTR_retriever [54].
Analysis of WGD events
In order to study the evolution of the H. tibetana genome, we compared the protein sequences from H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis and grape using BLASTP (p-value ≤ 1E − 07). MCScanX was used to detect syntenic blocks between genomes [55]. The synonymous substitution rate (Ks) values of gene pairs across syntenic blocks were calculated using KaKs_Calculator 2.0 [56]. GO and KEGG enrichment analyses were performed using clusterProfiler (v. 3.14.0) [57].
Comparative analysis of H. tibetana, H. rhamnoides subsp. mongolia and H. rhamnoides subsp. sinensis genome
Using the MUMmer (v 3.23) software, we detected collinearity between H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis genomes [58]. An analysis of SNPs and indels was conducted with MUMmer (v 3.23) to compare H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis genome. The first step of this alignment was to use the parameter “-maxmatch -c 100 -l50” to align H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. sinensis. Then, “delta-filter -1” parameter was used to filter the alignment results. As described in previous studies, MUMmer (v 3.23a) was used to examine SV between H. tibetana, H. rhamnoides subsp. mongolia, and H. rhamnoides subsp. Sinensis [59]. As described by Zhang et al., SV was detected in the exons, promoters, and downstream regions of the H. tibetana genome [60].
RNA extraction, sequencing, and analysis
Samples of root, stem, leave, and fruit tissues from three individuals were collected for total RNA extraction. We extracted total RNA from each sample using the MiniBEST Plant RNA Extraction Kit (TaKaRa, China) following the manufacturer’s protocol. The cDNA was synthesized using RNA to cDNA EcoDry Premix (Clontech) with oligo (dT) primer. The paired-end library was constructed based on the Paired-End sample Preparation kit protocol (Illumina, USA). RNA sequencing was performed on the NextSeq 500 platform (Illumina, USA). For RNA-seq analysis, we first used the HISAT2 software package to map RNA-seq data of roots, stems, leaves, and fruits to the H. tibetana genome [61]. The RSEM software was used to calculate the gene expression level as fragments per kilobase per million (FPKM) [62]. We used DESeq2 to identify differential expression genes with an adjusted p-value ≤ 0.05.
Identification of genes involved in the flavonoid pathway
In order to identify the candidate genes related to the flavonoid biosynthesis in the H. tibetana genome, we mapped the genes involved in the flavonoid biosynthesis pathway in A. thaliana to H. tibetana genome using blastp. Genes with E-value < 1E − 5 were identified as candidate genes related to flavonoid biosynthesis. The MEGA 7 software was used to construct ML phylogenetic trees using the homologous protein sequences from the four species [51].
Construction of transforming vectors and transformation of sea buckthorn
Gene-specific primers were used to clone the full-length open reading frames (ORFs) of HtC4H1 and HtC4H2 from young leaf cDNA of H. tibetana. Primers for the HtC4H1 and HtC4H2 genes containing Kpn I and Sal I cleavage sites were designed, and the HtC4H1 and HtC4H2 fragment was cloned from the extracted plasmid as the target fragment. The target fragment was inserted into the pCAMBIA1302-EGFP + 2 × P35s vector using a Seamless Cloning Kit (Beyotime, China) and transformed into an agrobacterium K599 competent cell (Huayueyang, China). Positive monoclonal colonies were selected for PCR verification and incubated with a TY liquid medium. When the OD600 value was about 0.4–0.6, the bacterial solution was injected into the stem base of sea buckthorn seedlings as described previously [63]. After growing in soil for 2 months, the roots with green fluorescence in sea buckthorn seedlings were collected.
Determination of flavonoid content of transgenic hairy roots and qRT-PCR analysis
The Plant Flavonoid Content Assay Kit (Solarbio, China) was used to determine flavonoid content in transgenic hairy roots. A UV spectrophotometer was used to measure the absorbance of samples at 420 nm. As described above, the RNA was extracted from approximately 0.1 g of transgenic hairy roots and analyzed quantitatively using real-time PCR. Total RNA from transgenic hairy roots and normal hairy roots was extracted using the MiniBEST Plant RNA Extraction Kit (TaKaRa). A PrimeScript™ RT reagent Kit (TaKaRa, Dalian, China) was used to synthesize cDNA. With gene-specific primers (Table S8), we performed quantitative real-time RT-PCR (qRT-PCR) with TB Green® Fast qPCR Mix (TaKaRa, Dalian, China) on the ABI StepOnePlus Real-Time PCR System (Applied Biosystems). The conditions for thermal cycling were 50 °C for 2 min and 95 °C for 20 s and run at 95 °C for 5 s and 60 °C for 20 s for 40 cycles [33]. Relative mRNA levels were calculated using the 2−ΔΔCt method. The sea buckthorn 18S gene was used to normalize the relative expression of genes. In all experiments, three biological replicates were examined.
Availability of data and materials
The genome assembly of the H. tibetana is available on the NCBI website (NCBI) under accession number PRJNA1070417 [64]. The raw sequencing data used for de novo whole-genome assembly and transcriptome data of Illumina RNA-seq are available from the BioProject under accession number PRJNA1070417 [64].
Abbreviations
- QTP:
-
Qinghai-Tibetan Plateau
- Mya:
-
Million years ago
- PAL:
-
Phenylalanine ammonia lyase
- C4H:
-
Cinnamic acid hydroxylase
- 4CL:
-
4-Coumaric acid CoA ligase
- CHS:
-
Chalcone synthase
- CHI:
-
Chalcone isomerase
- FNSI and FNS II:
-
Flavone synthase I and II
- F3′5′H:
-
Flavonol 3′5′-hy-droxylase
- FLS:
-
Flavonol synthase
- DFR:
-
Dihydroflavonol 4-reductase
- TEs:
-
Transposable elements
- CEGMA:
-
Core eukaryotic gene mapping approach
- LTRs:
-
Long-terminal repeats
- TDC:
-
Tryptophan decarboxylase
- CIPK:
-
CBL-interacting protein kinase
- FAH2:
-
Fatty acid 2-hydroxylase 1-like
- DIR:
-
Dirigent protein-like
- WGD:
-
Whole-genome duplication
- POLD3:
-
DNA-directed DNA polymerase
- XRCC2:
-
X-ray repair cross complementing 2
- SV:
-
Structural variation
- APEX2:
-
AP endonuclease 2 protein
- POLD2:
-
DNA polymerase delta subunit 2
- CIPK:
-
CBL-interacting protein kinase
- ORFs:
-
Pen reading frames
- qRT-PCR:
-
Quantitative real-time RT-PCR
- DEGs:
-
Differentially expressed genes
References
Xing Y, Ree RH. Uplift-driven diversification in the Hengduan Mountains, a temperate biodiversity hotspot. Proc Natl Acad Sci U S A. 2017;114(17):E3444–51.
Jia D, Liu T, Wang L, Zhou D, Liu J. Evolutionary history of an alpine shrub Hippophae tibetana (Elaeagnaceae): allopatric divergence and regional expansion. Biol J Linn Soc Lond. 2011;102:37–50.
He S, Cao W, Chen Y. The uplift of Qinghai-Xizang (Tibet) Plateau and the vicariance speciation of glyptosternoid fishes (Siluriformes: Sisoridae). Sci China C Life Sci. 2001;44(6):644–51.
Zhu W, Jiang N, Chen G, Zhang D, Zheng Z, Fan D. Divergent shifts and responses of plant autumn phenology to climate change on the Qinghai-Tibetan Plateau. Agric For Meteorol. 2017;239:166–75.
Zhang Q, Kong D, Shi P, Singh VP, Sun P. Vegetation phenology on the Qinghai-Tibetan Plateau and its response to climate change (1982–2013). Agric For Meteorol. 2018;248:408–17.
Wen J, Zhang JQ, Nie ZL, Zhong Y, Sun H. Evolutionary diversifications of plants on the Qinghai-Tibetan Plateau. Front Genet. 2014;5(4):1–16.
Cheviron ZA, Brumfield RT. Genomic insights into adaptation to high-altitude environments. Heredity (Edinb). 2012;108(4):354–61.
Qiu Q, Zhang G, Ma T, Qian W, Wang J, Ye Z, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44(8):946–51.
Li Y, Wu DD, Boyko AR, Wang GD, Wu SF, Irwin DM, et al. Population variation revealed high-altitude adaptation of Tibetan mastiffs. Mol Biol Evol. 2014;31(5):1200–5.
Zeng X, Long H, Wang Z, Zhao S, Tang Y, Huang Z, et al. The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau. Proc Natl Acad Sci U S A. 2015;112(4):1095–100.
Kalendar R, Tanskanen J, Immonen S, Nevo E, Schulman AH. Genome evolution of wild barley (Hordeum spontaneum)by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc Natl Acad Sci U S A. 2000;97(12):6603–7.
Zhang J, Tian Y, Yan L, Zhang G, Wang X, Zeng Y, et al. Genome of plant maca (Lepidium meyenii) illuminates genomic basis for high-altitude adaptation in the Central Andes. Mol Plant. 2016;9(7):1066–77.
Guo X, Hu Q, Hao G, Wang X, Zhang D, Ma T, et al. The genomes of two Eutrema species provide insight into plant adaptation to high altitudes. DNA Res. 2018;25(3):307–15.
Sun K, Chen X, Ma R, Li C, Wang Q, Ge S. Molecular phylogenetics of Hippophae L. (Elaeagnaceae) based on the internal transcribed spacer (ITS) sequences of nrDNA. Plant Syst Evol. 2002;235(1):121–34.
Zhou X, Tian L, Zhang J, Ma L, Li X, Tian C. Rhizospheric fungi and their link with the nitrogen-fixing Frankia harbored in host plant Hippophae rhamnoides L. J Basic Microbiol. 2017;57(12):1055–64.
Wang H, Laqiong, Sun K, Lu F, Wang Y, Song Z, et al. Phylogeographic structure of Hippophae tibetana (Elaeagnaceae) highlights the highest microrefugia and the rapid uplift of the Qinghai-Tibetan Plateau. Mol Ecol. 2010;19(14):2964–79.
Cao J, Zheng Y, Xia X, Wang Q, Xiao J. Total flavonoid contents, antioxidant potential and acetylcholinesterase inhibition activity of the extracts from 15 ferns in China. Ind Crops Prod. 2015;75:135–40.
Ciesarova Z, Murkovic M, Cejpek K, Kreps F, Tobolkova B, Koplik R, et al. Why is sea buckthorn (Hippophae rhamnoides L.) so exceptional? A review. Food Res Int. 2020;133(109170):1–18.
Pop RM, Socaciu C, Pintea A, Buzoianu AD, Sanders MG, Gruppen H, et al. UHPLC/PDA-ESI/MS analysis of the main berry and leaf flavonol glycosides from different Carpathian Hippophae rhamnoides L. varieties. Phytochem Anal. 2013;24(5):484–92.
Yu L, Diao S, Zhang G, Yu J, Zhang T, Luo H, et al. Genome sequence and population genomics provide insights into chromosomal evolution and phytochemical innovation of Hippophae rhamnoides. Plant Biotechnol J. 2022;20(7):1257–73.
Tohge T, de Souza LP, Fernie AR. Current understanding of the pathways of flavonoid biosynthesis in model and crop plants. J Exp Bot. 2017;68(15):4013–28.
Kersey PJ. Plant genome sequences: past, present, future. Curr Opin Plant Biol. 2019;48:1–8.
Edwards D, Batley J. Plant genome sequencing: applications for crop improvement. Plant Biotechnol J. 2010;8(1):2–9.
Wu Z, Chen H, Pan Y, Feng H, Fang D, Yang J, et al. Genome of Hippophae rhamnoides provides insights into a conserved molecular mechanism in actinorhizal and rhizobial symbioses. New Phytol. 2022;235(1):276–91.
Fu X, Wu J, Ma X, Li K, Zhang H, Wu S, et al. The Chromosome-level genome of Elaeagnus moorcroftii Wall., an economically and ecologically important tree species in drylands. Diversity (Basel). 2022;14(468):1–11.
Zhang T, Qiao Q, Novikova PY, Wang Q, Yue J, Guan Y, et al. Genome of Crucihimalaya himalaica, a close relative of Arabidopsis, shows ecological adaptation to high altitude. Proc Natl Acad Sci U S A. 2019;116(14):7137–46.
Guo W, Xin M, Wang Z, Yao Y, Hu Z, Song W, et al. Origin and adaptation to high altitude of Tibetan semi-wild wheat. Nat Commun. 2020;11(1):5085.
Xing L, Hu P, Liu J, Witek K, Zhou S, Xu J, et al. Pm21 from Haynaldia villosa encodes a CC-NBS-LRR protein conferring powdery mildew resistance in wheat. Mol Plant. 2018;11(6):874–8.
Yeh YH, Panzeri D, Kadota Y, Huang YC, Huang PY, Tao CN, et al. The Arabidopsis malectin-like/LRR-RLK IOS1 is critical for BAK1-dependent and BAK1-independent pattern-triggered immunity. Plant Cell. 2016;28(7):1701–21.
Xie P, Liu J, Lu R, Zhang Y, Sun X. Molecular evolution of the Pi-d2 gene conferring resistance to rice blast in Oryza. Front Genet. 2022;13(991900):1–14.
Jiang Z, Ge S, Xing L, Han D, Kang Z, Zhang G, et al. RLP1.1, a novel wheat receptor-like protein gene, is involved in the defence response against Puccinia striiformis f. sp. tritici. J Exp Bot. 2013;64(12):3735–46.
Liu S, Zhang X, Xiao S, Ma J, Shi W, Qin T, et al. A single-nucleotide mutation in a GLUTAMATE RECEPTOR-LIKE gene confers resistance to Fusarium wilt in Gossypium hirsutum. Adv Sci (Weinh). 2021;8(7):2002723.
Karlson CKS, Mohd Noor SN, Khalid N, Tan BC. CRISPRi-mediated down-regulation of the cinnamate-4-hydroxylase (C4H) gene enhances the flavonoid biosynthesis in Nicotiana tabacum. Biology (Basel). 2022;11(1127):1–14.
Zou K, Liu X, Zhang D, Yang Q, Fu S, Meng D, et al. Flavonoid biosynthesis is likely more susceptible to elevation and tree age than other branch pathways involved in phenylpropanoid biosynthesis in Ginkgo leaves. Front Plant Sci. 2019;10(983):1–17.
Hou S, Du W, Hao Y, Han Y, Li H, Liu L, et al. Elucidation of the regulatory network of flavonoid biosynthesis by profiling the metabolome and transcriptome in tartary buckwheat. J Agric Food Chem. 2021;69(25):7218–29.
Kim YS, Kim YB, Kim Y, Lee MY, Park SU. Overexpression of cinnamate 4-hydroxylase and 4-coumaroyl CoA ligase prompted flavone accumulation in Scutellaria baicalensis hairy roots. Nat Prod Commun. 2014;9(6):803–7.
Yao X, Lu Z, Song Y, Hu X, Corlett RT. A chromosome-scale genome assembly for the holly (Ilex polyneura) provides insights into genomic adaptations to elevation in Southwest China. Hortic Res. 2022;9:uhab049.
Sheng K, Sun Y, Liu M, Cao Y, Han Y, Li C, et al. A reference-grade genome assembly for Gossypium bickii and insights into its genome evolution and formation of pigment glands and gossypol. Plant Commun. 2022;3(100421):1–15.
Ma B, Wu J, Shi TL, Yang YY, Wang WB, Zheng Y, et al. Lilac (Syringa oblata) genome provides insights into its evolution and molecular mechanism of petal color change. Commun Biol. 2022;5(686):1–13.
Cheng H, Concepcion GT, Feng X, Zhang H, Li H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 2021;18(2):170–5.
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80.
Zhang X, Zhang S, Zhao Q, Ming R, Tang H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nat Plants. 2019;5(8):833–45.
Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016;3(1):99–101.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60.
Tarailo-Graovac M, Chen N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr Protoc Bioinformatics. 2009;Chapter 4:Unit 4.10.
Benson G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999;27(2):573–80.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78.
Chan PP, Lowe TM. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 2019;1962:1–14.
Nawrocki EP. Annotating functional RNAs in genomes using Infernal. Methods Mol Biol. 2014;1097:163–97.
Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinformatics. 2011;Chapter 6:Unit 6.12.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Lu YY, Tang K, Ren J, Fuhrman JA, Waterman MS, Sun F. CAFE: aCcelerated Alignment-FrEe sequence analysis. Nucleic Acids Res. 2017;45(W1):W554–9.
Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 2007;35(Web Server issue):W265–8.
Ou S, Jiang N. LTR_retriever: a highly accurate and sensitive program for identification of long terminal repeat retrotransposons. Plant Physiol. 2018;176(2):1410–22.
Wang Y, Tang H, Debarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49.
Wang D, Zhang Y, Zhang Z, Zhu J, Yu J. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics. 2010;8(1):77–80.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Marcais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: a fast and versatile genome alignment system. PLoS Comput Biol. 2018;14(1):e1005944.
Delcher AL, Phillippy A, Carlton J, Salzberg SL. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002;30(11):2478–83.
Zhang J, Lei Y, Wang B, Li S, Yu S, Wang Y, et al. The high-quality genome of diploid strawberry (Fragaria nilgerrensis) provides new insights into anthocyanin accumulation. Plant Biotechnol J. 2020;18(9):1908–24.
Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–15.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12(323):1–16.
Meng D, Yang Q, Dong B, Song Z, Niu L, Wang L, et al. Development of an efficient root transgenic system for pigeon pea and its application to other important economically plants. Plant Biotechnol J. 2019;17(9):1804–13.
Zhang G. Genome sequencing of Hippophae tibetana. NCBI BioProject accession: PRJNA1070417. 2024. Accessed 29 Jan 2024.
Acknowledgements
The platform of the facilities from the State Key Laboratory of Tree Genetics and Breeding is acknowledged. We thank Jiayi Li for her helpful suggestion.
Funding
This work was supported by the National Natural Science Foundation of China (32101554) and The National Key R&D Program of China (2022YFD2200401-5).
Author information
Authors and Affiliations
Contributions
JZ and CH conceived the project and were responsible for the project initiation. JZ and CH supervised and managed the project and research. JW collected the samples. Experiments and analyses were designed by GZ, YS, and NC. Data generation and bioinformatic analyses were led by GZ. The manuscript was organized, written, and revised by GZ, JZ, and CH. All authors read and revised the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
k-mer distribution of the H. tibetana genome. (a) k-mer = 17 Depth and k-mer number frequency distribution. (b) k-mer = 17 depth and k-mer type frequency distribution. Figure S2. The Venn diagram of gene function annotations in H. tibetana obtained using four databases, including InterPro, Swiss-Prot, NR, and KEGG. Figure S3. Gene family and KEGG enrichment analysis of unique genes in H. tibetana. (A) GO terms enriched in unique genes of H. tibetana. (B) KEGG pathways enriched in unique genes of H. tibetana. Figure S4. All differentially expressed genes (DEGs) between different tissues. Figure S5. All DEGs were classified into four clusters according to their expression patterns.
Additional file 2: Table S1.
Statistics of H. tibetana assembled chromosomes. Table S2. Statistics of predicted protein-coding genes in H.tibetana. Table S3. Statistical information of non-coding RNA in H.tibetana genome. Table S4. Evaluation of genome assembly completeness with 1614 BUSCO groups. Table S5. Statistics of repetitive sequences in the H. tibetana genome. Table S6. Genes involved in the flavonoid pathway. Table S7. Expression level of genes in H. tibetana. Table S8. Primers used for qRT-PCR.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, G., Song, Y., Chen, N. et al. Chromosome-level genome assembly of Hippophae tibetana provides insights into high-altitude adaptation and flavonoid biosynthesis. BMC Biol 22, 82 (2024). https://doi.org/10.1186/s12915-024-01875-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-024-01875-4