
Abstract
Background
RNA splicing plays significant roles in fundamental biological activities. However, our knowledge about the roles of alternative splicing and underlying mechanisms during spermatogenesis is limited.
Results
Here, we report that Serine/arginine-rich splicing factor 2 (SRSF2), also known as SC35, plays critical roles in alternative splicing and male reproduction. Male germ cell-specific deletion of Srsf2 by Stra8-Cre caused complete infertility and defective spermatogenesis. Further analyses revealed that deletion of Srsf2 disrupted differentiation and meiosis initiation of spermatogonia. Mechanistically, by combining RNA-seq data with LACE-seq data, we showed that SRSF2 regulatory networks play critical roles in several major events including reproductive development, spermatogenesis, meiotic cell cycle, synapse organization, DNA recombination, chromosome segregation, and male sex differentiation. Furthermore, SRSF2 affected expression and alternative splicing of Stra8, Stag3 and Atr encoding critical factors for spermatogenesis in a direct manner.
Conclusions
Taken together, our results demonstrate that SRSF2 has important functions in spermatogenesis and male fertility by regulating alternative splicing.
Similar content being viewed by others

Background
Spermatogenesis is a consistent and highly organized developmental process by which male germline stem cells divide and differentiate to produce mature spermatozoa. In mammalian testes, this process consists of three phases: mitosis, meiosis and spermiogenesis [1]. In the first phase of spermatogenesis, mitosis is characterized by the self-renewal and differentiation of spermatogonial stem cells (SSCs), which are also known as Asingle (As) spermatogonia. There are two outlets for As spermatogonia, self-renewal to maintain the germline stem cell pool and differentiation to enter meiosis after multiple rounds of mitotic divisions of undifferentiated spermatogonia [2]. As spermatogonia undergo unconventional mitotic processes to produce Apaired (Apr) spermatogonia and Aaligned (Aal) spermatogonia [3]. These spermatogonial progenitors including committed As, Apr, and Aal spermatogonia, are uniformly identified as undifferentiated spermatogonia. Then, Aal spermatogonia transform into type A1 spermatogonia and further go through a series of mitoses to form A2, A3, A4, intermediate (In) and B spermatogonia. These germ cells are called differentiating spermatogonia [4]. Next, B spermatogonia will divide into the pre-leptotene stage to prepare for entering meiosis which is initiated by retinoic acid (RA) and STRA8 [5, 6]. Any mistake in the proliferation and differentiation of SSCs can lead to failure of spermatogenesis, further resulting in severe consequences including infertility [7].
Alternative splicing (AS) is one of the most important transcriptional and post-transcriptional regulatory mechanisms to enrich the amount of mRNA and protein isoforms from a single gene, and these different protein isoforms always have different structural characteristics and functions [8,9,10]. Generally, AS occurs more frequently in highly complex organs and organisms [11,12,13]. There are numerous AS events during many developmental processes. Recently, it has been shown that several proteins including RAN-Binding Protein 9 (RANBP9), PTB protein 2 (Ptbp2), MORF-related gene on chromosome 15 (MRG15) and Breast carcinoma amplified sequence 2 (BCAS2) play important roles in AS events during spermatogenesis [14,15,16,17], indicating the importance of AS events during spermatogenesis, however, the functional significance of AS in the testis remains ambiguous, and the roles and regulation of AS in spermatogenesis are very limited.
The serine/arginine-rich splicing factors (SRs) have an exceedingly critical role in the alternative splicing process of precursor RNAs. The SRs can identify the splicing components of precursor RNA, then recruit and assemble spliceosomes to promote or inhibit the occurrence of alternative splicing events [18]. There is a substantial amount of researches indicating that SRs are involved in nearly every step of spliceosome assembly, genomic stability, mRNA export, mRNA stability and translation [19, 20]. Serine/arginine-rich splicing factor 2 (SRSF2), also known as SC35, is a member of the SRs protein family. It is an essential element of the nuclear structure, speckles [21]. Recently, several studies have suggested that SRSF2 plays important roles in regulating gene transcription, mRNA stability, genomic stability, and translation [22,23,24,25]. Also, some findings suggested that SRSF2 may serve as a therapeutic target for various diseases [26,27,28,29]. SRSF2 is also expressed in testis, however, its functions in male germ cells are still completely unknown.
Here, by crossing Srsf2Floxed/Floxed (Srsf2F/F) mice with Stra8-Cre mice to generate mutant mice with specific deletion of the Srsf2 gene in male germ cells, we found that the SRSF2 knockout caused complete infertility and germ cells were drastically lost during spermatogenesis. Further investigation revealed that deletion of the Srsf2 gene in germ cells affected the differentiation of spermatogonia and meiosis initiation. By combining advanced linear amplification of complementary DNA ends and sequencing (LACE-seq) and RNA-seq with bioinformatics analysis, we unbiasedly mapped the binding sites of SRSF2 at single-nucleotide resolution and revealed the changes of the transcriptome and transcripts splicing in SRSF2-null testes. Our results showed that SRSF2 deletion caused abnormal alternative splicing during spermatogenesis. In particular, we found that SRSF2 directly regulated the expressions of Stra8, Stag3 and Atr via AS, which have pivotal roles during spermatogenesis.
Results
SRSF2 is essential for male fertility
To investigate the function of SRSF2 in spermatogenesis, we first analyzed the expression of SRSF2 in the testis by using the anti-SRSF2 antibody. As a well-known marker of nuclear speckles, staining of cross-sections of seminiferous tubules in the adult mouse testis showed that SRSF2 was expressed in both germ cells and somatic cells of the testis (Fig. 1A), suggesting that SRSF2 may play a potential role in spermatogenesis.

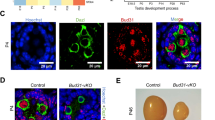
SRSF2 is essential for male fertility. A Representative images of localization of SRSF2 (green) and MVH (red) in the control and Srsf2cKO testes of 8-week-old mice. The DNA was stained with DAPI. Scale bar: (top) 50 μm; (bottom) 20 μm. B Schematic diagram of deletion of Srsf2 exons 1 and 2 and generation of Srsf2 Δ allele by Stra8 Cre-mediated recombination in male germ cells. C Genotyping PCRs were performed using Srsf2 flox and Srsf2 delta primers. D Quantitative RT-PCR analyses showing Srsf2 mRNA level was decreased. β-actin was used as the internal control. E Western blotting analysis of SRSF2 protein in Srsf2WT and Srsf2cKO total testes of 8-week-old mice. β-actin was detected as an internal control. F Pregnancy rates (%) of plugged wild-type females after mating with Srsf2WT and Srsf2cKO 8-week-old males. G Average litter size of plugged wild-type females after mating with Srsf2WT and Srsf2.cKO 8-week-old males. For this part, 3 mice (8-week-old) of each genotype were used for the analysis. Data are presented as the mean ± SEM. P < 0.05(*), 0.01(**) or 0.001(***)
Then, we generated Srsf2 conditional knockout mice (referred to as Srsf2cKO) by crossing Srsf2Floxed/Floxed (Srsf2F/F) mice in which the first and second exons were floxed [30], and Stra8-Cre mice in which cre activity is initiated at 3 days after birth [31]. Srsf2 was specifically deleted (Fig. 1B and C), and the knockout efficiency of SRSF2 was confirmed by using quantitative RT-PCR and Western blotting. The mRNA and protein level of SRSF2 was significantly decreased in testes of Srsf2cKO mice (Fig. 1D and E). Thus, we successfully established male germ cell-specific knockout mice for SRSF2. The breeding assays showed that the Srsf2cKO male mice were completely infertile (Fig. 1F and G). Although copulatory plugs were routinely observed, no pups were obtained when adult Srsf2cKO males were mated with normal fertile females.
Srsf2 depletion causes abnormal spermatogenesis in cKO mice
To determine the reasons of infertility in Srsf2cKO male mice, we firstly performed histological analyses. Compared with controls, the testes of Srsf2cKO mice were much smaller (Fig. 2A). The testis weight and the testis weight to body weight ratio of Srsf2cKO mice was significantly lower (Fig. 2B and C). Then we analyzed the histology of the epididymes and testes by Hematoxylin and Eosin (H&E) staining. The results showed that no mature spermatozoa were found in the epididymal lumens of Srsf2cKO mice (Fig. 2D). The seminiferous tubules of Srsf2WT testes contained a basal population of spermatogonia, several types of spermatocytes and spermatids. However, germ cells were severely reduced in number, spermatocytes and spermatids were absent in the seminiferous tubules of Srsf2cKO testes (Fig. 2E). These results indicated that germ cell-specific Srsf2 knockout results in spermatogenesis failure and thus male infertility.

SRSF2 is required for spermatogenesis. A The testes of Srsf2cKO were smaller than those of the control (8-week-old, the same as below). B Testis weight of Srsf2WT and Srsf2cKO 8-week-old male mice (n = 3). C Testis weight to body weight ratio of Srsf2WT and Srsf2cKO 8-week-old male mice (n = 3). Data are presented as the mean ± SEM. P < 0.05(*), 0.01(**) or 0.001(***). D Histological analysis of the caudal epididymes of the Srsf2WT and Srsf2cKO mice. (Scale bar: 50 μm) (E) Histological analysis of the seminiferous tubules of the Srsf2WT and Srsf2.cKO mice. Scale bar: (top) 100 μm; (bottom) 50 μm. For this part, 3 mice (8-week-old) of each genotype were used for the analysis. Data are presented as the mean ± SEM. P < 0.05(*), 0.01(**) or 0.001(***)
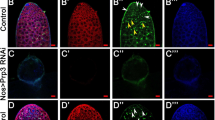
To validate the above results, we performed immunofluorescent staining by using lectin peanut agglutinin (PNA) and antibodies against SOX9 and MVH, markers for the acrosomes of spermatids, Sertoli cells, and germ cells, respectively. Immunofluorescence results indicated that there were no PNA-positive signals in the seminiferous tubules of Srsf2cKO testes and the number of MVH positive signals was significantly reduced in cKO testicular sections compared with those in the control (Fig. 3A). Sertoli cells marker SOX9 staining showed that the number and location of Sertoli cells did not show an obvious change (Fig. 3A).

Srsf2 deficient germ cells fail to progress into meiosis. A PNA-lectin histochemistry (green), SOX9 (a marker of Sertoli cells, white) and MVH (a marker of germ cells, red) immunofluorescence analysis of the Srsf2WT and Srsf2cKO 8-week-old male mice. Scale bar: (top) 50 μm; (bottom) 20 μm. B γH2AX (green) and SYCP3 (red) immunofluorescence analysis of the Srsf2WT and Srsf2cKO 8-week-old male mice. Scale bar: (top) 50 μm; (bottom) 20 μm. C PLZF (green) and MVH (red) immunofluorescence analysis of the Srsf2WT and Srsf2cKO male mice at P6, P8, P10 and P12. Scale bar, 20 μm. In this part, 3 mice of each genotype were used for the analysis
Meiotic recombination and homologous chromosome synapsis are two pivotal events in meiotic progression. Next we examined meiotic progression by immunostaining the axial element component of the synaptonemal complex with SYCP3 and double-strand break (DSB) marker γH2AX. Similarly, immunofluorescence results indicated that there were no SYCP3 positive signals in the seminiferous tubules of Srsf2cKO testes at 8-week-old and P12, suggesting that meiosis initiation is disrupted after SRSF2 cKO (Fig. 3B and Additional file 1: Fig. S1).
To further identify which stage of spermatogenesis was impaired in SRSF2-deficient mice, we performed immunofluorescence staining of the undifferentiated spermatogonia marker promyelocytic leukaemia zincfinger protein (PLZF; also known as Zbtb16) and the germ cell marker MVH (mouse vasa homologue) to characterize the first wave of spermatogenesis in mice at postnatal day 6 (P6), P8, P10, and P12. The results showed that nearly all the germ cells were undifferentiated spermatogonia in both the Srsf2WT and Srsf2cKO group at P6 (Fig. 3C). Then the undifferentiated spermatogonia proliferated to self-renew or divided into differentiating spermatogonia from P8 to P12 in the Srsf2WT group. However, MVH positive signals and PLZF positive signals were always nearly co-localized in the Srsf2cKO group from P6 to P12 (Fig. 3C), showing that nearly all the germ cells were undifferentiated spermatogonia in Srsf2cKO group. Altogether, these results indicated that the differentiation of spermatogonia was affected in Srsf2cKO mice, which may further cause the failure of meiosis initiation.
Changes in transcriptome and splicing of transcripts in SRSF2-null testes
According to the above-presented data, SRSF2 cKO mice displayed defects in spermatogenesis. To investigate a comprehensive perspective of the mechanisms of SRSF2 deletion in male germ cells, we isolated mRNA from Srsf2WT and Srsf2cKO testes at P10 and then performed RNA sequencing (RNA-seq). RNA-seq results firstly showed the reduction of Srsf2 RNA in Srsf2cKO mice testes (Fig. 4A). Clustering and principal component analysis (PCA) clearly distinguished the gene expression patterns of Srsf2cKO mice testes from the Srsf2WT mice testes (Fig. 4B). A total of 977 genes were upregulated, and 1742 genes were downregulated in Srsf2cKO testes (FDR of < 0.05, |log2FoldChange|≥ 0.6) (Fig. 4C). Heatmap showed hierarchical clustering of differential expression genes (DEGs) of Srsf2WT and Srsf2cKO testes (Fig. 4D). To obtain more comprehensive information, we then performed Gene Ontology (GO) annotation. GO analysis showed that these upregulated genes were involved in reproductive development, sex differentiation, and gonad development (Fig. 4E). Meiotic cell cycle, chromosome segregation, DNA repair, DNA recombination, and cellular processes involved in reproduction in multicellular organisms were significantly enriched among these downregulated genes (Fig. 4E). In short, these differential expression genes may account for the SRSF2-null phenotypes in spermatogenesis.

Transcriptome and splicing of transcripts changes in SRSF2-null testes. A RNA-seq results showing the reduction of Srsf2 RNA in Srsf2cKO mice testes. Three independent RNA-seq experiments are shown. B Srsf2cKO groups rather than to Srsf2WT groups are clustered together by PCA. C Volcano plot showing transcriptome changes between Srsf2WT and Srsf2cKO testes. D Heatmap showing hierarchical clustering of differential expression genes of Srsf2WT and Srsf2cKO male mice testes. E GO term enrichment analysis of upregulated genes and downregulated genes. F The five different types of alternative splicing (AS) events. The numbers of abnormal AS events were counted between Srsf2WT and Srsf2cKO testes by rMATS software. In this part, 3 mice of each genotype were used for the analysis
Because SRSF2 played critical roles in regulating RNA splicing, we then analyzed the five different types of AS events between Srsf2WT and Srsf2cKO testes by using the rMATS computational tool. Compared with the Srsf2WT group, a total of 320 AS events were identified as significantly changed in the Srsf2cKO group (|Diff|> 0.05, FDR < 0.001). Among these 320 changed AS events, most (267) of AS events were skipped exons (SE). Moreover, there were 17 alternative 3′ splice sites (A3SS), 15 alternative 5′ splice sites (A5SS), 16 mutually exclusive exons (MXE), and 5 retained introns (RI) (Fig. 4F and Additional file 2: Fig. S2). Together, these results suggested that SRSF2 is essential for RNA splicing during spermatogenesis.
Binding landscape of SRSF2 proteins analysis in mouse testes
To further investigate the molecular mechanisms by which SRSF2 causes the failure of spermatogenesis, we performed LACE-seq analysis by using testes at P10 to profile SRSF2-binding sites in testes (Fig. 5A). IgG served as the negative control in our works. Then, we removed the nonspecific background by excluding the overlapping peaks shown in the IgG controls. Two independent replicates with a high correlation in read counts were pooled together for the following analysis (Fig. 5B). Among these SRSF2 clusters, more than half of them were derived from intergenic regions, while others were aligned to intron, CDS (coding sequence), UTR3 (3′ untranslated region), and UTR5 (5′ untranslated region) (Fig. 5C). We also found that SRSF2 “preferentially” bound to exons and enriched between 0 and 100 nt of the 5′ and 3′ exonic sequences flanking the constitutive splice sites as revealed by analyzing the distributions of SRSF2-binding peaks within 500 nucleotides (nt) upstream or downstream of the constitutive splice site (Fig. 5D). Among these SRSF2 peaks, most of them had at least one CG-rich hexamer, and more than half of the peaks contained at least one top-10 motif (Fig. 5E and Additional file 6: Table S2). GO analysis showed that these SRSF2-binding genes were involved in the regulation of RNA splicing, reproductive development, male sex differentiation, regulation of synapse organization, and regulation of chromosome segregation (Fig. 5F). Then, SRSF2-specific targets incorporated in Fig. 5F were analyzed. The association network shows the subgroups based on the function of involved genes (Fig. 5G). Together, these analyses suggested that SRSF2 is essential for reproductive development.

Global landscape of SRSF2-binding sites in mouse testes as revealed by using LACE-seq. A Flowchart of the LACE-seq method. RBP, represents RNA-binding protein. A circled B represents biotin modification. N, represents random nucleotide; V represents A, G or C. IVT, represents in vitro transcription. B Spearman correlation plot between SRSF2 LACE-seq replicates in total testes for assessing the reproducibility of the data. Spearman correlation for the reads counts of each sample was calculated from two replicates. C Genomic distribution of SRSF2 binding sites in testes. CDS, coding sequence. UTR3, 3′ untranslated region. UTR5, 5′ untranslated region. D Schematic analysis showing the distribution of SRSF2-binding sites in the vicinity of the 5′ exon–intron and the 3′ intron–exon boundaries (500 nt upstream and 500 nt downstream of 3′SS; 500 nt upstream and 500 nt downstream of 5′SS). E SRSF2-binding motifs identified by LACE-seq in mouse testes. F GO enrichment map of SRSF2-binding genes. G Network analysis of the enriched GO terms of SRSF2-specific targets
SRSF2 affects expression and AS of Stra8, Stag3 and Atr in a direct manner
By combining RNA-seq data with LACE-seq identified peaks, we identified 262 downregulated, and 187 upregulated transcripts as direct targets of SRSF2 in testes (Additional file 7: Table S3). To obtain more comprehensive information, similarly, we then performed GO annotation. GO analysis showed that both significantly upregulated genes and SRSF2-binding genes were involved in reproductive development, male sex differentiation, and germ cell development (Fig. 6A). Then, these significantly upregulated genes and SRSF2-specific targets incorporated in Fig. 6A were analyzed. The association network shows the subgroups based on the function of involved genes (Fig. 6B). And spermatogenesis, meiotic cell cycle, male gamete generation, chromosome segregation, DNA repair, and DNA recombination were significantly enriched among these both significantly downregulated genes and SRSF2-binding genes (Fig. 6C). Then, these significantly downregulated genes and SRSF2-specific targets incorporated in Fig. 6C were analyzed. The association network shows the subgroups based on the function of involved genes (Fig. 6D). We next validated these both significantly DEGs and SRSF2-binding genes which were involved in spermatogenesis by using quantitative polymerase chain reaction (qPCR) to check the mRNA abundance (Fig. 6E and F). These data reflected that deletion of SRSF2 directly affects the expression levels of critical genes involved in spermatogenesis.

The expressions of key SRSF2-binding genes involved in the spermatogenesis change after Srsf2 KO. A Correlation analysis between the RNA-seq and LACE-seq. GO analysis of the significantly upregulated genes and SRSF2-binding genes. B Network analysis of the enriched GO terms of the significantly upregulated genes and SRSF2-specific targets. C Correlation analysis between the RNA-seq and LACE-seq. GO analysis of the significantly downregulated genes and SRSF2-binding genes. D Network analysis of the enriched GO terms of the significantly downregulated genes and SRSF2-specific targets. E Quantitative RT-PCR validation of the expression of genes involved in (B). β-actin was used as the internal control. Data are presented as the mean ± SEM. P < 0.05(*), 0.01(**) or 0.001(***). F Quantitative RT-PCR validation of the expression of genes involved in (D). β-actin was used as the internal control. Data are presented as the mean ± SEM. P < 0.05(*), 0.01(**) or 0.001(***)
Furthermore, we investigated the relationship of SRSF2-binding genes, DEGs, and AS genes to confirm the direct targets that account for the abnormal spermatogenesis after SRSF2 cKO. Venn diagram showed that 14 SRSF2 directly binding genes were differentially down-regulated and spliced (Fig. 7A). These genes included Stra8, Stag3, Atr, Hmga1, and Setx, and all of them were necessary for the male germ cell development (Fig. 7B). We then researched SRSF2 regulatory mechanism on the expression of Stra8, Stag3 and Atr by combining the RNA-seq with LACE-seq. The data showed that the abundance of Stra8 mRNA was decreased and the ratio of exon 2 skipping was increased after SRSF2 cKO. Similarly, the abundance of Stag3 mRNA was decreased and the ratio of exon 19 and 20 skipping was increased after SRSF2 cKO. The abundance of Atr mRNA was decreased and the ratio of exon 34 skipping was increased after SRSF2 cKO (Fig. 7C). We also performed RT-PCR, semiquantitative reverse transcription PCR and sequencing technology to confirm the above results (Fig. 7D, E and Additional file 4: Fig. S4A, B, C). These experiments indicated that SRSF2 affects the expression levels and AS of Stra8, Stag3 and Atr in a direct manner, which were critical for male germ cell differentiation and development.

SRSF2 affects expression and alternative splicing of Stra8, Stag3 and Atr in a direct manner. A Venn diagram shows the correlation among SRSF2-binding genes, DEGs, and AS genes. B The detailed genes of SRSF2-binding, differentially expressed, and AS. C A magnified view showing RNA-seq and LACE-seq signals of the selected candidate genes. IgG, immunoglobulin G. D Quantitative RT-PCR validation of the expression of Stra8, Stag3, and Atr. E Semiquantitative RT-PCR analysis of AS patterns of the changed spliced genes in Srsf2WT and Srsf2cKO testes at P10 (n = 4 per group). PCR primers are listed in Additional file 5: Table S1. The scheme and cumulative data on percentage of the indicated fragments are shown accordingly
Discussion
As members of the serine arginine-rich protein family, SRs which include 12 members in mammalian (SRSF1–12) are well-known for their regulatory function of splicing [32]. The first SRs identified were SRSF1 (previously known as SF2/ASF) and SRSF2 (previously known as SC35) [33]. SRs consist of one or two RNA-recognition motifs (RRM) in the N-terminus and arginine/serine amino acid sequences (RS domain) in the C-terminus [34]. In general, RRM can recognize RNA and determine the binding of SRs to RNA, while the RS domain can regulate diverse protein-RNA and protein–protein interactions [33]. Like other SR splicing factors, several studies in recent years have suggested that SRSF2 have important roles in regulating gene transcription, mRNA stability, genomic stability, and translation [22,23,24,25]. Also, some findings suggested that SRSF2 may serve as a therapeutic target for various diseases [26,27,28,29].
Recently, it has been found that RNA-binding proteins (RBPs) have important functions during germline and early embryo development. As a RBP, SRSF2 is also expressed in testis, however, its functions in male germ cells is still completely unknown. In this study, by crossing Srsf2F/F mice with Stra8-Cre mice to generate mutant mice, we found that SRSF2 is essential for spermatogenesis and fertility in males.
The RBPs could serve post-transcriptional functions to determine cellular RNA and protein levels. For the past few years, high throughput sequencing techniques have become an increasingly essential tool for biological research. RNA immunoprecipitation with sequencing (RIP-seq) and crosslinking immunoprecipitation coupled with high-throughput sequencing (CLIP-seq or HITS-CLIP) are two major methods to identify RBPs targets from millions of cells [35, 36]. There are also some modified versions, such as iCLIP, irCLIP and eCLIP [37,38,39]. Up to now, LACE-seq is the latest method developed by us, which can unbiasedly map the binding sites of these RBPs at single-nucleotide resolution in low-input cells [40]. To gain a comprehensive perspective of the mechanisms of SRSF2 depletion in male germ cells, we isolated testes from wildtype mouse at P10 and systematically profiled binding landscape of SRSF2 proteins by using LACE-seq. The results showed that SRSF2 proteins could bind numerous genes in a direct manner. Then, our analysis showed that these SRSF2-binding genes were closely involved in the regulation of RNA splicing, reproductive development, male sex differentiation, regulation of synapse organization, and regulation of chromosome segregation. In addition, RNA-seq analysis further showed that transcriptome and splicing of transcripts change in SRSF2-null testes. By combining RNA-seq and LACE-seq data, we identified 262 downregulated, and 187 upregulated transcripts as direct targets of SRSF2 in testes. The two omics data reflected that deletion of SRSF2 directly affects the expression levels of critical genes involved in spermatogenesis, such as Sycp1, Rnf114, Setx, Hmgb2, Gata4, Sox8, Amh, Kitl, and Axl.
Retinoic acid (RA) is an important factor of spermatogenesis, with functions on spermatogonial differentiation and subsequently initiation of meiosis [41, 42]. The two certain targets for RA are Stra8 and Kit. Several surveys indicated that Stra8 has two different roles during spermatogenesis. On one hand, under the influence of RA, Stra8 functions as a transcriptional repressor of the pluripotency program during differentiation of spermatogonia. When differentiating spermatogonia are near the end of their mitotic phase, Stra8 switches to the second role and acts as a transcription activator of genes involved in meiosis initiation [43,44,45]. In addition to RA signaling, Dazl is also regarded as a regulator of meiotic initiation [46]. Of particular note, in Srsf2cKO mice, the differentiation of spermatogonia and meiosis initiation were disrupted. Except for Stra8, Stag3 and Atr are crucial regulators of meiotic processes during spermatogenesis [47,48,49,50,51]. The two omics data also indicated that SRSF2 affects the expression levels and AS of Stra8, Stag3 and Atr in a direct manner, which are critical for the male germ cell development process. Also, we found that the reduced expression and abnormal AS of Dazl were indirectly cuased by SRSF2 deletion (Additional file 3: Fig.S3 and Additional file 4: Fig. S4D). Nonsense-mediated mRNA decay (NMD) is an RNA surveillance mechanism that detects the mRNAs harboring premature termination codons or truncated mRNA. Thus, the abnormal AS events of pre-mRNAs may be degraded by NMD pathway and results in the altered expression of these target genes. Through sequence analysis, we found that the skipping of exon 19- 20 in Stag3 mRNA and the skipping of exon 34 in Atr mRNA caused a frameshift and introduced premature termination codon (PTC) (Fig. 7E, Additional file 4: Fig. S4B and C). But the skipping of exon 2 in Stra8 mRNA and the skipping of exon 8 in Dazl mRNA did not cause a frameshift and PTC (Fig. 7E, Additional file 4: Fig. S4A and D).
Conclusions
In summary, our study has demonstrated for the first time that SRSF2 has important functions in male fertility and spermatogenesis, especially in the differentiation of spermatogonia and meiosis initiation. Mechanistic analyses reveal that SRSF2 is essential for posttranscriptional regulation by specifically adjusting the gene expression and AS in direct or indirect manners during spermatogenesis. These abnormally expressed genes, such as Stra8, Stag3, Atr and Dazl, caused by SRSF2 deletion finally result in the failure of spermatogenesis and male infertility.
Methods
Mice
Mice lacking Srsf2 in male germ cells (referred to as Srsf2cKO) were generated by crossing Srsf2Floxed/Floxed (Srsf2F/F) mice with Stra8-Cre mice. All transgenic mouse lines have C57BL/6 J genomic background. Genotyping PCR for Srsf2 was performed using the following primers: forward: GTTATTTGGCCAAGAATCACA, and reverse: TAGCCAGTTGCTTGTTCCAA. The PCR conditions were as follows: 94 ℃ for 5 min; 35 rounds of 94 ℃ for 30 s, 60 ℃ for 30 s, and 72 ℃ for 30 s; and 72 ℃ for 5 min. Genotyping PCR for Stra8-Cre was performed using the following primers: forward: ACTCCAAGCACTGGGCAGAA, wildtype reverse: GCCACCATAGCAGCATCAAA and reverse: CGTTTACGTCGCCGTCCAG. The PCR conditions were as follows: 94 ℃ for 5 min; 35 rounds of 94 ℃ for 30 s, 60 ℃ for 30 s, and 72 ℃ for 30 s; and 72 ℃ for 5 min. Four genotypes in the progeny, including Srsf2F/+, Srsf2F/−, Srsf2F/+; Stra8-Cre and Srsf2F/−; Stra8-Cre were identified. The Srsf2F/+ male mice were used as control group.
The mice were maintained under specific-pathogen-free (SPF) conditions and housed under controlled environmental conditions with free access to water and food. All animal operations were approved by the Animal Care and Use Committee of the Institute of Zoology, Chinese Academy of Sciences (CAS).
Antibodies
Β-actin antibody (mouse, sc-47778; Santa Cruz); SYCP3 (mouse, sc-74569; Santa Cruz); γH2AX (rabbit, 9718; Cell Signaling Technology, Inc.); MVH (mouse, ab27591; Abcam); SOX9 antibody (rabbit, AB5535, Sigma-Aldrich); PLZF antibody (goat, AF2944, R&D Systems); SFRS2 polyclonal antibody (rabbit, 20,371–1-AP, Proteintech); SC35 antibody (mouse, S4045, Sigma-Aldrich); green-fluorescent Alexa Fluor® 488 conjugate of lectin PNA (L21409, Thermo). Horseradish peroxidase–conjugated secondary antibodies were purchased from Zhongshan Golden Bridge Biotechnology Co, LTD (Beijing). Alexa Fluor 488–conjugated antibody, 594–conjugated antibody and Alexa Fluor 647–conjugated antibody were purchased from Life Technologies.
Breeding assay
Males of different genotypes (8 weeks) were used for the breeding assay. Each male mouse was caged with two wild-type ICR (Institute of Cancer Research) females (7 weeks), and their vaginal plugs were checked every morning. The number of pups in each cage was counted within a week of birth. Each male underwent six cycles of the above breeding assay.
Immunoblotting
To prepare protein extracts, testes were homogenized in RIPA lysis buffer supplemented with protease and phosphatase inhibitor cocktail (Roche Diagnostics). After transient ultrasound treatment, the testis lysates were incubated on ice for 30 min and then centrifuged at 4 ℃, 12,000 rpm for 20 min. The supernatant was transferred to a new tube and quantified using a BCA reagent kit (Beyotime, P0012-1). Then equal volume loading buffer was added. After being boiled at 95 ℃ for 10 min, the protein lysates were used for immunoblotting analysis. Immunoblotting was performed as described previously [52]. Briefly, the separated proteins in SDS-PAGE were electrically transferred to a polyvinylidene fluoride membrane. After incubation with primary and secondary antibodies, the membranes were scanned with Bio-Rad ChemiDoc XRS + .
Tissue collection and histological analysis
For histological analysis, at least three adult mice for each genotype were analyzed. Testes and caudal epididymides were dissected immediately following euthanasia. The tissues were then fixed in Bouin’s fixative (saturated picric acid: 37% formaldehyde: glacial acetic acid = 15: 5: 1) overnight at room temperature, dehydrated in an ethanol series, and embedded in paraffin wax. Then, 5 μm sections were cut with a microtome. After 48 ℃ overnight drying, the sections were deparaffinized in xylene, hydrated by a graded alcohol series and stained with Hematoxylin and Eosin for histological analysis. Images were collected with a Nikon inverted microscope with a charge coupled device (CCD) (Nikon, Eclipse Ti-S, Tokyo, Japan).
Immunofluorescence
Testes used for immunostaining were fixed in 4% paraformaldehyde (pH 7.4) overnight at 4 ℃, dehydrated, and embedded in paraffin. Paraffin-embedded testes were cut into sections of 5 μm thickness. Then, the sections were deparaffinized, immersed in sodium citrate buffer (pH 6.0) and heated for 15 min in a microwave for antigen retrieval. After blocking with 5% donkey serum albumin, sections were incubated with primary antibodies at 4 ℃ overnight. Then the sections were incubated with an appropriate FITC-conjugated secondary antibody. The nuclei were stained with DAPI. Images were captured using a laser scanning confocal microscope LSM880 (Carl Zeiss, Germany).
RNA extraction and gene expression analysis
Total RNA was extracted from whole testes using TRNzol Universal Reagent (cat. # DP424, Tiangen, China) according to the manufacturer’s instructions. Then reverse transcription (RT) was performed using the 5X All-In-One RT MasterMix (cat. # G490, Abm, Canada). RT-PCR was performed using the UltraSYBR Mixture (cat. # CW0957, Cowin Bio, China) on a LightCycler 480 instrument (Roche). The results were analyzed based on the 2−ΔΔCt method to calculate the fold changes. β-actin was used as an internal control. At least three independent experiments were analyzed. All primer sequences are listed in the Additional file 5: Table S1.
Semiquantitative PCR experiment was carried out with primers (listed in Additional file 5: Table S1) amplifying endogenous transcripts. Then the PCR products were detected on 2% agarose gels. Gapdh was used as an internal control.
RNA sequencing and data analysis
Total testes samples were used from P10 Srsf2WT and Srsf2cKO male mice according to three individual collections. One Total RNA was extracted from whole testes using TRNzol Universal Reagent (cat. # DP424, Tiangen, China) according to the manufacturer’s instructions. The quality of RNA samples was examined by NanoDrop 2000&8000 and Agilent 2100 Bioanalyzer, Agilent RNA 6000 Nano Kit. The high-quality RNAs were used to prepare the libraries, followed by high-throughput sequencing on an Illumina NovaSeq 6000. The RNA sequencing experiment was supported by Annoroad BioLabs.
After trimming adaptor sequence and rRNA, the retained reads from Srsf2 control and cKO samples were aligned to mouse genome (mm9) using HISAT2 with default parameters. Only non-RCR duplicate and uniquely mapped reads were used for subsequent analysis. Significantly changed genes were screened using DESeq2 with |log2FC|> 0.6 and FDR < 0.05. Alternative splicing events were identified by rMATS with default parameters. Only events with FDR < 0.001 and splicing difference > 0.05 were regarded as significant.
LACE-sequencing and data analysis
Total testes samples were used from P10 WT male mice for LACE-seq. LACE-seq method was performed as described recently by us [40]. Briefly, the samples were firstly irradiated twice with UV-C light on ice at 400 mJ. Then RNA immunoprecipitation of the samples was performed. The immunoprecipitated RNAs were then fragmented by MNase and dephosphorylated. Then a series of steps were performed to include, reverse transcription, first-strand cDNA capture by streptavidin beads, poly(A) tailing, pre-PCR, IVT, RNA purification, RT, PCR barcoding and deep sequencing.
The adapter sequences and poly(A) tails at the 3′ end of raw reads were removed using Cutadapt (v.1.15) with two parameters: -f fastq -q 30,0 -a ATCTCGTATGCCGTCTTCTGCTT -m 18 –max-n 0.25 –trim-n., and -f fastq -a A -m 18 -n 2. Clean reads were first aligned to mouse pre-rRNA using Bowtie, and the remaining unmapped reads were then aligned to the human (hg19) or mouse (mm9) reference genome. For LACE-seq data mapping, two mismatches were allowed (Bowtie parameters: -v 2 -m 10 –best -strata; -v 2 -k 10 –best -strata). Peaks were identified by Piranha with parameters: -s -b 20 -p 0.01. Peaks without IgG signal were selected for further usage. For motif analysis, LACE-seq peaks/clusters were first extended 30 nt to 5′ upstream, and overrepresented hexamers in the extended sequences were identified as previously described [53]. The consensus motifs were generated from the top-10 enriched hexamers using WebLogo.
Statistical analysis
All of the experiments were performed at least three times independently. Paired two-tailed Student’s t-test was used for statistical analysis. Data analyses were carried out via GraphPad Prism 8.00 (GraphPad Software, Inc.) and presented as mean ± SEM and P < 0.05(*), 0.01(**) or 0.001(***) was considered statistically significant.
Availability of data and materials
All data generated or analysed during this study are included in this published article, its supplementary information files and publicly available repositories. The RNA-seq data and LACE-seq data were deposited in GEO (https:// www. ncbi. nlm. nih. gov/ geo/) under accession number GSE206537. The individual data values for Figs. 1, 2, 6, and 7, as well as Additional file 3: Fig. S3 are provided in Additional file 9.
Abbreviations
- A3SS:
-
Alternative 3′ splice sites
- A5SS:
-
Alternative 5′ splice sites
- AS:
-
Alternative splicing
- BCAS2:
-
Breast carcinoma amplified sequence 2
- CLIP-seq:
-
Crosslinking immunoprecipitation coupled with high-throughput sequencing
- DEGs:
-
Differential expression genes
- DSB:
-
Double-strand break
- GO:
-
Gene Ontology
- H&E:
-
Hematoxylin and Eosin
- LACE-seq:
-
Linear amplification of complementary DNA ends and sequencing
- MRG15:
-
MORF-related gene on chromosome 15
- MVH:
-
Mouse vasa homologue
- MXE:
-
Mutually exclusive exons
- NMD:
-
Nonsense-mediated mRNA decay
- PLZF:
-
Promyelocytic leukaemia zincfinger protein
- PNA:
-
Peanut agglutinin
- Ptbp2:
-
PTB protein 2
- RA:
-
Retinoic acid
- RANBP9:
-
RAN-Binding Protein 9
- RBPs:
-
RNA-binding proteins
- RI:
-
Retained introns
- RIP-seq:
-
RNA immunoprecipitation with sequencing
- RRM:
-
RNA-recognition motifs
- SSCs:
-
Spermatogonial stem cells
References
Oatley JM, Brinster RL. Regulation of spermatogonial stem cell self-renewal in mammals. Annu Rev Cell Dev Biol. 2008;24:263–86.
Yang QE, Oatley JM. Spermatogonial stem cell functions in physiological and pathological conditions. Curr Top Dev Biol. 2014;107:235–67.
Kanatsu-Shinohara M, Shinohara T. Spermatogonial stem cell self-renewal and development. Annu Rev Cell Dev Biol. 2013;29:163–87.
Song HW, Wilkinson MF. Transcriptional control of spermatogonial maintenance and differentiation. Semin Cell Dev Biol. 2014;30:14–26.
Hogarth CA, Griswold MD. The key role of vitamin A in spermatogenesis. J Clin Invest. 2010;120(4):956–62.
Zhou Q, Nie R, Li Y, Friel P, Mitchell D, Hess RA, et al. Expression of stimulated by retinoic acid gene 8 (Stra8) in spermatogenic cells induced by retinoic acid: an in vivo study in vitamin A-sufficient postnatal murine testes. Biol Reprod. 2008;79(1):35–42.
Nagaoka SI, Hassold TJ, Hunt PA. Human aneuploidy: mechanisms and new insights into an age-old problem. Nat Rev Genet. 2012;13(7):493–504.
Lee Y, Rio DC. Mechanisms and Regulation of Alternative Pre-mRNA Splicing. Annu Rev Biochem. 2015;84:291–323.
Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463(7280):457–63.
Song H, Wang L, Chen D, Li F. The function of pre-mRNA alternative splicing in mammal spermatogenesis. Int J Biol Sci. 2020;16(1):38–48.
Merkin J, Russell C, Chen P, Burge CB. Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Sci. 2012;338(6114):1593–9.
Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456(7221):470–6.
Li Q, Li T, Xiao X, Ahmad DW, Zhang N, Li H, et al. Specific expression and alternative splicing of mouse genes during spermatogenesis. Molecular Omics. 2020;16(3):258–67.
Liu W, Wang F, Xu Q, Shi J, Zhang X, Lu X, et al. BCAS2 is involved in alternative mRNA splicing in spermatogonia and the transition to meiosis. Nat Commun. 2017;8(1):14182.
Iwamori N, Tominaga K, Sato T, Riehle K, Iwamori T, Ohkawa Y, et al. MRG15 is required for pre-mRNA splicing and spermatogenesis. Proc Natl Acad Sci. 2016;113(37):E5408–15.
Zagore LL, Grabinski SE, Sweet TJ, Hannigan MM, Sramkoski RM, Li Q, et al. RNA binding protein Ptbp2 is essential for male germ cell development. Mol Cell Biol. 2015;35(23):4030–42.
Barsh GS, Bao J, Tang C, Li J, Zhang Y, Bhetwal BP, et al. RAN-Binding Protein 9 is Involved in Alternative Splicing and is Critical for Male Germ Cell Development and Male Fertility. PLoS Genet. 2014;10(12):e1004825.
Zheng X, Peng Q, Wang L, Zhang X, Huang L, Wang J, et al. Serine/arginine-rich splicing factors: the bridge linking alternative splicing and cancer. Int J Biol Sci. 2020;16(13):2442–53.
Huang Y, Gattoni R, Stévenin J, Steitz JA. SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell. 2003;11(3):837–43.
Savisaar R, Hurst LD. Purifying selection on exonic splice enhancers in intronless genes. Mol Biol Evol. 2016;33(6):1396–418.
Li K, Wang Z. Splicing factor SRSF2-centric gene regulation. Int J Biol Sci. 2021;17(7):1708–15.
Qian W, Iqbal K, Grundke-Iqbal I, Gong CX, Liu F. Splicing factor SC35 promotes tau expression through stabilization of its mRNA. FEBS Lett. 2011;585(6):875–80.
Xiao R, Sun Y, Ding JH, Lin S, Rose DW, Rosenfeld MG, et al. Splicing regulator SC35 is essential for genomic stability and cell proliferation during mammalian organogenesis. Mol Cell Biol. 2007;27(15):5393–402.
Wang Z, Li K, Chen W, Wang X, Huang Y, Wang W, et al. Modulation of SRSF2 expression reverses the exhaustion of TILs via the epigenetic regulation of immune checkpoint molecules. Cell Mol Life Sci. 2020;77(17):3441–52.
Wang Z, Liu Q, Lu J, Fan P, Xie W, Qiu W, et al. Serine/Arginine-rich splicing factor 2 modulates herpes simplex virus type 1 replication via regulating viral gene transcriptional activity and pre-mRNA splicing. J Biol Chem. 2016;291(51):26377–87.
Luo C, Cheng Y, Liu Y, Chen L, Liu L, Wei N, et al. SRSF2 regulates alternative splicing to drive hepatocellular carcinoma development. Cancer Res. 2017;77(5):1168–78.
Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V, et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood. 2012;120(15):3080–8.
Wu SJ, Kuo YY, Hou HA, Li LY, Tseng MH, Huang CF, et al. The clinical implication of SRSF2 mutation in patients with myelodysplastic syndrome and its stability during disease evolution. Blood. 2012;120(15):3106–11.
Lance A, Druhan LJ, Vestal CG, Steuerwald NM, Hamilton A, Smith M, et al. Altered expression of CSF3R splice variants impacts signal response and is associated with SRSF2 mutations. Leukemia. 2020;34(2):369–79.
Wang H-Y, Xu X, Ding J-H, Bermingham JR, Fu X-D. SC35 plays a role in T cell development and alternative splicing of CD45. Mol Cell. 2001;7(2):331–42.
Sadate-Ngatchou PI, Payne CJ, Dearth AT, Braun RE. Cre recombinase activity specific to postnatal, premeiotic male germ cells in transgenic mice. Genesis. 2008;46(12):738–42.
Busch A, Hertel KJ. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA. 2012;3(1):1–12.
Manley JL, Krainer AR. A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 2010;24(11):1073–4.
Jeong S. SR proteins: binders, regulators, and connectors of RNA. Mol Cells. 2017;40(1):1–9.
Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456(7221):464–9.
Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, et al. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell. 2010;40(6):939–53.
König J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17(7):909–15.
Zarnegar BJ, Flynn RA, Shen Y, Do BT, Chang HY, Khavari PA. irCLIP platform for efficient characterization of protein-RNA interactions. Nat Methods. 2016;13(6):489–92.
Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods. 2016;13(6):508–14.
Su R, Fan L-H, Cao C, Wang L, Du Z, Cai Z, et al. Global profiling of RNA-binding protein target sites by LACE-seq. Nat Cell Biol. 2021;23(6):664–75.
Huang HF, Hembree WC. Spermatogenic response to vitamin A in vitamin A deficient rats. Biol Reprod. 1979;21(4):891–904.
Koubova J, Menke DB, Zhou Q, Capel B, Griswold MD, Page DC. Retinoic acid regulates sex-specific timing of meiotic initiation in mice. Proc Natl Acad Sci U S A. 2006;103(8):2474–9.
Endo T, Romer KA, Anderson EL, Baltus AE, de Rooij DG, Page DC. Periodic retinoic acid-STRA8 signaling intersects with periodic germ-cell competencies to regulate spermatogenesis. Proc Natl Acad Sci U S A. 2015;112(18):E2347–56.
Ishiguro KI, Matsuura K, Tani N, Takeda N, Usuki S, Yamane M, et al. MEIOSIN Directs the Switch from Mitosis to Meiosis in Mammalian Germ Cells. Dev Cell. 2020;52(4):429–45 e1.
Sinha N, Whelan EC, Tobias JW, Avarbock M, Stefanovski D, Brinster RL. Roles of Stra8 and Tcerg1l in retinoic acid induced spermatogonial differentiation in mousedagger. Biol Reprod. 2021;105(2):503–18.
Lin Y, Gill ME, Koubova J, Page DC. Germ cell-intrinsic and -extrinsic factors govern meiotic initiation in mouse embryos. Sci. 2008;322(5908):1685–7.
Llano E, Gomez-H L, García-Tuñón I, Sánchez-Martín M, Caburet S, Barbero JL, et al. STAG3 is a strong candidate gene for male infertility. Hum Mol Genet. 2014;23(13):3421–31.
van der Bijl N, Ropke A, Biswas U, Woste M, Jessberger R, Kliesch S, et al. Mutations in the stromal antigen 3 (STAG3) gene cause male infertility due to meiotic arrest. Hum Reprod. 2019;34(11):2112–9.
Fukuda T, Fukuda N, Agostinho A, Hernandez-Hernandez A, Kouznetsova A, Hoog C. STAG3-mediated stabilization of REC8 cohesin complexes promotes chromosome synapsis during meiosis. EMBO J. 2014;33(11):1243–55.
Prieto I, Suja JA, Pezzi N, Kremer L, Martinez AC, Rufas JS, et al. Mammalian STAG3 is a cohesin specific to sister chromatid arms in meiosis I. Nat Cell Biol. 2001;3(8):761–6.
Widger A, Mahadevaiah SK, Lange J, ElInati E, Zohren J, Hirota T, et al. ATR is a multifunctional regulator of male mouse meiosis. Nat Commun. 2018;9(1):2621.
Lei WL, Han F, Hu MW, Liang QX, Meng TG, Zhou Q, et al. Protein phosphatase 6 is a key factor regulating spermatogenesis. Cell Death Differ. 2020;27(6):1952–64.
Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, et al. Genome-wide analysis of PTB-RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell. 2009;36(6):996–1006.
Acknowledgements
We thank Dr Xiang-Dong Fu for providing the Srsf2F/F mice. We appreciate and acknowledge Shiwen Li and Xili Zhu for their technical assistance. We thank all members of the Sun lab for their help and discussion.
Funding
This study was supported by National Key R&D Program of China (2021YFA1100302, 2019YFA0109900), National Natural Science Foundation of China (82301806), and Guangdong Basic and Applied Basic Research Foundation (2021A1515111118, 2020A1515011414).
Author information
Authors and Affiliations
Contributions
Conceptualization was performed by W.L., Q.S., W.Q., F.S., and Z.W.; methodology and investigation by W.L., Z.D., T.M., R.S., Y.L., W.L., Y.H., C.Z. and Y.G.; visualization by S.S., and M.L.; supervision by H.S. and Z.H.; writing—original draft by W.L., and Y.L.; and writing—review and editing by C.L., F.S., and Q.S. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All experiments were conducted following the guidelines and with the approval of the Animal Care and Use Committee of the Institute of Zoology, Chinese Academy of Sciences (CAS) (No. IOZ20160033).
Consent for publication
All contributors give consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Spermatogenesis fails to progress into meiosis in Srsf2 deficient germ cells at P12 γH2AX (green) and SYCP3 (red) immunofluorescence analysis of the Srsf2WT and Srsf2cKO male mice at P12. Scale bar: (top) 50 μm; (bottom) 20 μm.
Additional file 2: Fig. S2.
SRSF2 regulates mRNA alternative splicing in testes Five AS events significantly affected by deletion of SRSF2 in the testes at P10. The different types of alternatively spliced events were shown.
Additional file 3: Fig. S3.
SRSF2 indirectly regulates splicing and expression of Dazl. (A) A magnified view showing RNA-seq signals of the Dazl gene. (B) Quantitative RT-PCR validation of the expression of Dazl. (C) Semiquantitative RT-PCR analysis of AS patterns of the changed spliced genes in Srsf2WT and Srsf2cKO testes at P10 (n = 4 per group). PCR primers are listed in Additional file 5: Table S1. The scheme and cumulative data on percentage of the indicated fragment are shown accordingly.
Additional file 4: Fig. S4.
The effect of SRSF2 deletion-introduced exon skipping. (A) The effect of SRSF2 deletion-introduced exon 2 skipping on Stra8 mRNA. The sequence of targeted exon was showed between Srsf2WT and Srsf2cKOgroups. (B) The effect of SRSF2 deletion-introduced exon 19 and exon 20 skipping on Stag3 mRNA. The sequence of targeted exon was showed between Srsf2WT and Srsf2cKOgroups. (C) The effect of SRSF2 deletion-introduced exon 34 skipping on Atr mRNA. The sequence of targeted exon was showed between Srsf2WT and Srsf2cKO groups. (D) The effect of SRSF2 deletion-introduced exon 8 skipping on Dazl mRNA. The sequence of targeted exon was showed between Srsf2WT and Srsf2cKO groups.
Additional file 5:
Table S1. Primer List.
Additional file 6:
Table S2. The top-10 enriched hexamers.
Additional file 7:
Table S3. Binding genes and DEGs.
Additional file 8.
Images of the original, uncropped gels/blots.
Additional file 9.
Individual data values.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lei, WL., Du, Z., Meng, TG. et al. SRSF2 is required for mRNA splicing during spermatogenesis. BMC Biol 21, 231 (2023). https://doi.org/10.1186/s12915-023-01736-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-023-01736-6