
Abstract
Background
Using genetically modified plants as natural dispensers of insect pheromones may eventually become part of a novel strategy for integrated pest management.
Results
In the present study, we first characterized essential functional genes for sex pheromone biosynthesis in the rice stem borer Chilo suppressalis (Walker) by heterologous expression in Saccharomyces cerevisiae and Nicotiana benthamiana, including two desaturase genes CsupYPAQ and CsupKPSE and a reductase gene CsupFAR2. Subsequently, we co-expressed CsupYPAQ and CsupFAR2 together with the previously characterized moth desaturase Atr∆11 in N. benthamiana. This resulted in the production of (Z)-11-hexadecenol together with (Z)-11-hexadecenal, the major pheromone component of C. suppressalis. Both compounds were collected from the transformed N. benthamiana headspace volatiles using solid-phase microextraction. We finally added the expression of a yeast acetyltransferase gene ATF1 and could then confirm also (Z)-11-hexadecenyl acetate release from the plant.
Conclusions
Our results pave the way for stable transformation of plants to be used as biological pheromone sources in different pest control strategies.
Similar content being viewed by others


Background
Moths rely strongly on sex pheromones for mate communication. Synthetic pheromones have been used for monitoring, mass trapping, and mating disruption in integrated pest management (IPM) for several decades [1, 2] as environmentally friendly alternatives or complements to conventional insecticides. They are species-specific and non-toxic, and the risk of pests evolving resistance to their own pheromone is very low. Compared to current standard approaches to pheromone synthesis [3], the use of biological factories for pheromone production may have several advantages, allowing cost-efficient production of moderate to large quantities of pheromones with high purity and a minimum of waste. A series of proof-of-concept studies have clearly demonstrated the potential of producing moth pheromones in both plant and yeast factories [4,5,6,7] to replace conventionally produced pheromones in existing systems for pheromone-based pest control. However, under certain circumstances, it may be advantageous to cultivate plants that actually release the pheromone volatiles in the field rather than accumulate them for harvest. Such pheromone-releasing plants could hypothetically and depending on context either protect themselves against moth pests by causing mating disruption or be cultivated together with other harvest crops for the purpose of attraction or mating disruption.
Deciphering the molecular mechanism of pheromone biosynthesis in moth species can provide a functional gene pool for producing customized pheromones in heterologous systems. Approximately 75% of the identified moth pheromones belong to the so-called type I sex pheromones; C10–C18 alcohols, acetates, or aldehydes that are biosynthesized from palmitic and stearic acid by consecutive steps of fatty acyl desaturation, chain-shortening or chain-elongation, reduction, and final modification [8]. Specific enzymes are used for each catalytic step. Fatty acyl desaturases (FADs) are the first essential enzymes that introduce double bonds in specific positions of carbon chains with strict regioselectivity and stereoselectivity. Fatty acyl reductases (FARs), which are responsible for reducing fatty acyls to fatty alcohols, are essential for forming functional groups of the fatty acid derivatives [8]. The genes encoding the essential enzymes involved in pheromone fatty acyl desaturation and reduction have been functionally characterized in many moth species [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25]. Characterization of FAD and FAR genes with different substrate and product specificities remains important for the production of tailored moth pheromones in heterologous systems.
The fatty alcohols may be actual pheromone components, or they may be further functionalized by the conversion of fatty alcohols into esters [26] or aldehydes [27,28,29]. Although in vivo labeling experiments have confirmed these biological reactions, no enzymes catalyzing the reactions in the pheromone glands have been identified and cloned from any insect species. In order to produce acetate pheromones in plants, Ding et al. [5] explored the possibility of using an acetyltransferase gene EaDAcT cloned from burning bush, Euonymus alatus. Transient expression resulted in the production of (Z)-11-hexadecenyl acetate (Z11-16:OAc), but the efficiency was low. Subsequently, Ding et al. [30] characterized a yeast acetyltransferase gene ATF1 which efficiently acetylates insect pheromone alcohols into acetates in yeast. However, the activity of ATF1 in plants remains to be explored.
The rice stem borer, Chilo suppressalis (Walker) (Lepidoptera: Crambidae), boring the stems of their host plants, is an infamous rice pest in East Asia, India, and Indonesia, causing great production reduction in rice crops [31]. In the 1970s and 1980s, the sex pheromone of female C. suppressalis was identified as a mixture of (Z)-11-hexadecenal (Z11-16:Ald), (Z)-13-octadecenal (Z13-18:Ald) [32, 33], and (Z)-9-hexadecenal (Z9-16:Ald) at the ratio of 100:13:11 [34]. In the present study, we functionally characterized several candidate genes likely to be involved in pheromone biosynthesis in C. suppressalis by heterologous expression in yeast and plant platforms, using the transcriptome data reported by Xia et al. [35]. We produced N. benthamiana genetically modified for expression of the functionally characterized C. suppressalis ∆11 desaturase CsupYPAQ, the fatty acyl reductase CsupFAR2 and the yeast acetyltransferase ATF1 that released a mixture of (Z)-11-hexadecenol (Z11-16:OH), Z11-16:OAc, and Z11-16:Ald. Our study contributes additional genes to the pool of key enzymes available for biotechnological production of moth pheromones, and more importantly, it is a significant step forward in the construction of genetically modified plants to be used as natural dispensers of insect pheromones as part of IPM strategies for pest control.
Results
CsupYPAQ and CsupKPSE are the functional FAD genes involved in pheromone biosynthesis in Chilo suppressalis
The C. suppressalis FAD-like genes CsupYPAQ and CsupKPSE (following the nomenclature proposed by Knipple et al. [13] naming desaturase genes based on the composition of 4 amino acid residues at a signature motif) displayed 1038 nt and 1059 nt ORFs that translated into 346 and 353 aa-proteins, respectively. In a phylogenetic analysis of lepidopteran FADs, CsupYPAQ clustered into the ∆11/∆10/multifunctional FAD subfamily, whereas CsupKPSE fell into the ∆9 (C16>C18) FAD clade (Fig. 1).

Phylogenetic tree of fatty acyl desaturases (FADs). The FAD tree was constructed by using amino acid sequences of lepidopteran FADs. The FADs functionally characterized in previous studies are classified into six subfamilies: ∆9 FAD subfamilies usually involved in normal fatty acid metabolism with preference for C16 (C16>C18) or C18 (C18>C16); the ∆11/∆10/multifunctional FAD subfamily; the ∆9 FAD subfamily with preference for C14-C26, involved in pheromone biosynthesis; and the ∆14 and ∆5/∆6 FAD subfamilies that introduce double bonds into unusual positions in saturated fatty acids. The predicted FAD genes CsupYPAQ and CsupKPSE from C. suppressalis are marked by green and red-filled circles, respectively. Values indicated at the nodes are bootstrap values based on 1000 replicates and bootstrap values <50% are not shown
The functional expression of CsupYPAQ and CsupKPSE indicated their involvement in pheromone biosynthesis. GC/MS analysis of yeast fatty acids showed that the yeast expressing CsupYPAQ produced a high amount of (Z)-11-hexadecenoic acid (Z11-16:acid) (Fig. 2a), while yeast expressing CsupKPSE produced high amounts of oleic acid (Z9-18:acid) and (Z)-9-hexadecenoic acid (Z9-16:acid) (Fig. 2b). Compared to the yeast expression results, CsupYPAQ showed a similar function in N. benthamiana (Fig. 3a). However, CsupKPSE expressed in N. benthamiana did not produce observable extra Z9-18:acid but only a very high amount of Z9-16:acid (Fig. 3b). In the wild type N. benthamiana, none of the monounsaturated potential pheromone precursors was produced (Fig. 3c).

Heterologous expression of fatty acyl desaturase candidates from Chilo suppressalis in ∆ole1/elo1 Saccharomyces cerevisiae. GC/MS analysis of fatty acid methyl ester profiles of yeast expressing the CsupYPAQ or CsupKPSE. The compounds produced from the introduced desaturases are labeled in red italics. Methyl (Z)-10-heptadecenoate (Z10–17:Me) was supplemented as nutrition to all the incubations. Displayed chromatograms are representative examples of at least six replicates

Heterologous expression of fatty acyl desaturase candidates from Chilo suppressalis in Nicotiana benthamiana. GC/MS analysis of fatty acid methyl ester profiles of a plant leaves expressing CsupYPAQ, b plant leaves expressing CsupKPSE, and c wild-type leaves. Native compounds from plants are labeled in black and the compounds produced from the introduced desaturases are labeled in red italics. Displayed chromatograms are representative examples of at least six replicates
CsupFAR2 is the FAR involved in pheromone biosynthesis in Chilo suppressalis
The gene candidate CsupFAR2 encompassed an ORF of 1404 nt, which corresponded to a 468 aa-protein. The phylogenetic analysis showed that CsupFAR2 could be classified as a pgFAR (Fig. 4). The functional assays of CsupFAR2 performed in both yeast and plant expression platforms demonstrated that it reduced the C. suppressalis pheromone precursors Z11-16:acid, Z9-16:acid, and (Z)-13-octadecenoic acid (Z13-18:acid) to the corresponding fatty alcohols (Figs. 5 and 6). A co-expression construct of CsupYPAQ-CsupFAR2 was produced (ca. 3500 nt) with the Gal1 promoter upstream of CsupFAR2. The yeast that was not expressing CsupYPAQ was supplemented with Methyl (Z)-11-hexadecenoate (Z11-16:Me). The yeast with the transformed empty vectors of Gateway adjusted pYES52 (Fig. 5a, b) and pYEX-CHT (Fig. 5c, d) did not produce fatty alcohol, while the yeast expressing CsupFAR2 or CsupYPAQ-CsupFAR2 produced Z11-16:OH, (Z)-13-octadecenol (Z13-18:OH), and palmityl alcohol (16:OH) (Fig. 5e–g). Compared to the yeast expressing only CsupFAR2 and supplemented with Z11-16:Me, the yeast co-expressing CsupYPAQ-CsupFAR2 (without supplement) produced a higher amount of target compounds (Fig. 5g). In the negative control, the yeast expressing CsupYPAQ only did not produce fatty alcohols (Fig. 5h). Besides Z11-16:OH, Z13-18:OH, and 16:OH, no other fatty alcohol species were detected in the yeast (Fig. 5).

Phylogenetic tree of fatty acyl reductases (FARs). The tree is constructed from mammal, arthropod, and Lepidoptera FARs using amino acid sequences. The pgFAR clade, which contains previously functionally characterized FARs involved in moth pheromone biosynthesis, is shown in green and marked by a bracket. The predicted C. suppressalis fatty acyl reductase CsupFAR2 is marked by a triangle. Values indicated at the nodes are bootstrap values based on 1000 replicates and bootstrap values <50% are not shown.

Heterologous expression of the fatty acyl reductase candidate gene CsupFAR2 and the desaturase gene CsupYPAQ in Saccharomyces cerevisiae INVSc strain. CsupFAR2 is constructed in the galactose inducible vector pYES52 and CsupYPAQ-CsupFAR2 is constructed in the Cu2+ inducible vector pYEX-CHT. GC/MS analysis of fatty alcohol profiles of yeast harboring the a pYEX52 empty vector supplemented with Z11-16:Me, b pYES52 empty vector without supplement, c pYEX-CHT empty vector supplemented with Z11-16:Me, d pYEX-CHT empty vector without supplement, e CsupFAR2 in pYES52 supplemented with Z11-16:Me, f CsupFAR2 in pYES52 without supplement, g CsupYPAQ-CsupFAR2, and h CsupYPAQ in pYEX-CHT without supplement. Fatty alcohols produced from the introduced genes are shown in red and italics. Displayed chromatograms are representative examples of at least six replicates.

Heterologous expression of fatty acyl reductase candidate gene CsupFAR2 and fatty acyl desaturase genes CsupYPAQ and CsupKPSE in Nicotiana benthamiana. GC/MS analysis of fatty alcohol (a–e) and corresponding fatty acid (f, j) profiles of plant leaves expressing the a, f CsupFAR2, b, g CsupYPAQ-CsupFAR2, c, h CsupKPSE-CsupFAR2, d, i CsupYPAQ-CsupKPSE-CsupFAR2, and wild type (e and j). Fatty alcohols produced by the introduced genes are shown in red. Eighteen micrograms of Z8-13:OAc per gram fresh leaf was added during the extraction as internal standard. Displayed chromatograms are representative examples of at least six replicates
In the N. benthamiana expression system, CsupFAR2 was demonstrated to be active with a range of substrates including saturated, monounsaturated, and polyunsaturated fatty acids. The leaves expressing CsupFAR2 reduced a high amount of 16:acid and a minor amount of α-linolenic acid (Z9,Z12,Z15-18:acid) to the corresponding 16:OH and linolenyl alcohol (Z9,Z12,Z15-18:OH) (Fig. 6a, f). However, it did not reduce other plant-derived fatty acids such as linoleic acid (Z9,Z12-18:acid), roughanic acid (Z7,Z10,Z13-16:acid), Z9-18:acid, or other saturated fatty acids than 16:acid in detectable amounts. When CsupYPAQ was co-introduced with CsupFAR2, the leaves produced plenty of Z11-16:OH. This gene combination resulted in a tenfold higher amount of Z11-16:OH than 16:OH and Z13-18:OH (Fig. 6b). When CsupKPSE-CsupFAR2 was co-expressed in the plant, in addition to 16:OH and linolenyl alcohol, a minor amount of (Z)-9-hexadecenol (Z9-16:OH) was produced (less than 1/3 of 16:OH), similar to the proportion of the precursor Methyl (Z)-9-hexadecanoate (Z9-16:Me) relative to Methyl palmitate (16:Me) (Fig. 6c, h). The plant co-expressing multiple genes CsupYPAQ-CsupKPSE-CsupFAR2 showed similar alcohol production compared to the plant expressing CsupYPAQ-CsupFAR2 (Fig. 6b, d), but Z9-16:acid was only produced in the plants expressing the CsupKPSE (Fig. 6g, i). The wild-type plant did not produce any of the alcohols mentioned above (Fig. 6e).
No functional alcohol oxidase genes characterized from Chilo suppressalis
A homology-based search yielded five fatty alcohol oxidase/dehydrogenase candidates (CsupFAO_15570, CsupFAO_9572, CsupADH_10975, CsupADH_14583, CsupADH_17286) from C. suppressalis (Table 1). The fatty alcohol oxidase (FAO) gene candidates encompassed ORFs around 1900 nt and had the highest amino acid identity of 26.5% between CsupFAO_15570 and an FAO (XM_500864) from the yeast Yarrowia lipolytica. The alcohol dehydrogenase (ADH) gene candidates encompassed ORFs around 1100 nt and had the highest amino acid identity of 68.6% between CsupADH_14583 and an ADH (NP_741507) from the nematode Caenorhabditis elegans.
We then individually co-expressed the five FAO and ADH-like genes and an additional ADH gene from Helicoverpa zea HzeaADH7, which was previously reported as an ADH-highly like gene with PG abundant expression in pheromone glands [36], in N. benthamiana leaves together with CsupYPAQ and CsupFAR2 in various combinations (Additional file 1: Table S1).
However, no results indicating the expected activity were obtained with any of the FAO/ADH gene candidates. In the GC/MS analysis of the leaf extracts, only a small peak of Z11-16:Ald was detected in the leaf samples when CsupYPAQ, CsupKPSE, CsupFAR2, and Csup15570 were co-expressed in the plant (Fig. 7a) and the control leaf samples from co-expression of CsupYPAQ, CsupKPSE, and CsupFAR2 also contained an albeit very small but still significant amount of Z11-16:Ald (Fig. 7b). This implied the existence of an endogenous plant activity that works on Z11-16:OH for producing Z11-16:Ald.

Heterologous expression of fatty alcohol oxidase candidate Csup15570 with desaturase genes CsupYPAQ and CsupKPSE and reductase gene CsupFAR2 from Chilo suppressalis in Nicotiana benthamiana. GC/MS analysis of Z11-16 fatty alcohol and aldehyde from plant leaves expressing a CsupYPAQ-CsupKPSE-CsupFAR2-Csup15570 and b CsupYPAQ-CsupKPSE-CsupFAR2. Fatty alcohol and aldehyde are shown in red. Displayed chromatograms are representative examples of at least six replicates
Transiently genetically modified Nicotiana benthamiana releasing moth pheromones
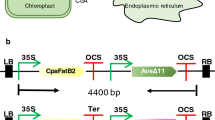
The newly identified desaturase and reductase genes with desired properties were used together with two genes previously identified, to produce plants transiently genetically modified to release moth pheromone compounds. Nicotiana benthamiana plants infiltrated with the functional ∆11 desaturases gene Atr∆11 and thioesterase gene CpuFatB1, and the newly identified desaturase and reductase genes CsupYPAQ and CsupFAR2 (Fig. 8a), released a mixture of Z11-16:OH and Z11-16:Ald that could be collected as volatiles from the plant leaves (Fig. 8c, d). Upon co-expression of the acetyltransferase gene ATF1, Z11-16:OAc was also released together with Z11-16:OH and Z11-16:Ald. In addition, to explore the possibility of increasing the amount of moth pheromone compounds released from plant leaf, a N. tabacum trichome-specific promoter pCYP71D16 was used to control the expression of acetyltransferase gene ATF1. Consequently, the plant released significantly higher amounts of Z11-16:OAc, as well as Z11-16:Ald and Z11-16:OH, compared to when ATF1 expression was controlled by the constitutive promoter p35S (Fig. 8d). The accumulation of Z11-16:Ald and Z11-16:OH was also higher in the leaves when ATF1 was controlled by pCYP71D16, while Z11-16:OAc accumulation in the leaves showed no significant difference between the two strategies (Fig. 8e).

Rapid assembly of pheromone biosynthetic pathway in Nicotiana benthamiana for the production and release of moth sex pheromone components. The Cauliflower mosaic virus 35S promoter (p35S) and Octopine Synthase gene terminator (tOCS) have been used to regulate gene expression in plants. ATF1 has also been controlled by trichome-specific promoter pCYP71D16. a Step-wise metabolic engineering strategy for leaf-based pheromone production of (Z)-11-hexadecenol, (Z)-11-hexadecenal, and (Z)-11-hexadecenyl acetate. b Solid-phase microextraction (SPME) of headspace volatiles from a genetically modified N. benthamiana leaf 4 days after infiltration. c GC-MS chromatograms (INNOWax column) of volatiles collected during 24 h from an infiltrated N. benthamiana leaf. d The amount of collected pheromone compounds from released volatiles. e The amount of pheromone compounds from leaf extracts. The error bars represent the standard deviation (SD). N=6. Bars with the same letters represent treatments that are not significantly different at the P=0.05 level (two-way ANOVA followed by t-tests)
Discussion
Enabling plants to release heterologously produced moth pheromones is an essential prerequisite for developing an IPM strategy in which the “pheromone-releasing” plants can be used for mating disruption of pests or to selectively attract a specific pest insect. We successfully made N. benthamiana release the major pheromone compound, Z11-16:Ald, of C. suppressalis and the common pheromone compounds Z11-16:OH and Z11-16:OAc, by transient expression of the functionally characterized C. suppressalis ∆11 desaturase CsupYPAQ, the fatty acyl reductase CsupFAR2, and the yeast acetyltransferase gene ATF1. Furthermore, characterization of the highly active pheromone biosynthetic genes from C. suppressalis is an important addition to the genetic toolbox for constructing heterologous platforms to produce customized insect pheromones.
Understanding the mechanisms underpinning moth pheromone biosynthesis is considered the basis for developing biological factories for moth pheromone production. In the present study, we demonstrate the key roles of three genes in the biosynthesis of the C. suppressalis sex pheromone, i.e., a ∆11 desaturase CsupYPAQ that acts specifically on palmitic acid to produce Z11-16:acid; a ∆9 desaturase CsupKPSE producing Z9-16:acid, the precursor of the minor pheromone component from palmitic acid; and CsupFAR2 that transforms the acid precursors into the corresponding fatty alcohols. The enzyme(s) transforming the alcohols into the final aldehyde pheromone components remains elusive (Fig. 9).

Biosynthetic pathways towards the sex pheromone of Chilo suppressalis and the key genes involved in each step
In insects, the acyl-CoA desaturase gene family has had a very dynamic evolutionary history [13, 37, 38]. As a result of gene duplication bursts, many opportunities have arisen for evolution to explore protein space and produce proteins with unique and novel functions. In phylogenetic analysis, CsupYPAQ clusters in the Lepidoptera-specific XXXQ/E clade (the proteins contain an XXXQ/E amino acid motif 13 amino acids in front of the conserved histidine box of the desaturase sequences) (Fig. 1), which is a lineage that is composed of pheromone biosynthetic desaturases with diverse specificities [13]. With the exception of the ∆9 desaturase from the tortricid species Cydia pomonella [39], the FADs clustering in this clade are ∆11/∆10 FADs but in some cases, they have additional functions and are then classified as multifunctional. For example, Trichoplusia ni and Spodoptera exigua from Noctuoidae using ∆11 [12] and ∆11/∆12 [22] desaturations, respectively; Thaumetopoea pityocampa from Notodontidae using ∆11/∆13 desaturations [20]; Bombyx mori from Bombycidae using ∆11/10,12 desaturations [40]; Epiphyas postvittana, Choristoneura rosaceana, and Agryrotaenia velutinana from Tortricidae using Δ11 desaturations [11, 14, 15], from the same family Planotortrix octo using Δ10 desaturation [10] (Fig. 1). Interestingly, even though CsupYPAQ clusters close to OfuZ/E11 (Ostrinia furnacalis) and OnuZ/E11 (O. nubilalis) from crambid species, which produce both Z and E isomers of ∆11-tetradecenoic acid [18], it specifically produces (Z)-11-hexadecenoic acid. In addition, CsupYPAQ shares 59.7% and 61.8% of amino acid identity to SexiDes5 (S. exigua) and SlitDes5 (S. litura), respectively, which are also both ∆11 FADs and have a very wide substrate preference, from 14 to 18 carbon chain length fatty acids [22]. All evidence at hand advocates for caution when predicting gene functions based on homology to currently characterized gene sequences, and functional testing is essential in terms of enhancing our mechanistic understanding of the relation between sequence and function of desaturases. Ding et al. [30] reported that one amino acid (258E) at the cytosolic carboxyl terminus of the protein is critical for the Z activity of the C. rosaceana FAD. The relative specificity of CsupYPAQ makes it an interesting candidate to further explore the correlation between the sequence and function of FADs by using site-directed mutagenesis and functional testing. Functional assays of CsupYPAQ showed similar results in the yeast and plant platforms but CsupKPSE expression did not yield oleic acid in N. benthamiana while it did in S. cerevisiae. Phylogenetically, CsupKPSE is clustered in the “∆9, C16>C18” clade; however, it showed C18>C16 function when oleic acid was absent in the ole1/elo1 yeast.
Fatty acyl reductases are used for converting fatty acyls into their corresponding alcohols during moth pheromone biosynthesis. Sometimes, these FARs are referred to with the epithet “pheromone-gland specific” (pgFAR) although specific expression in the pheromone gland is not evident. Phylogenetically, the pgFAR clade containing the genes seems to be specific to Lepidoptera [8]. To date, several genes encoding pgFARs have been characterized in moth species such as Bombyx mori [24], O. nubilalis [23], Helicoverpa armigera [41], and Spodoptera spp. [25]. In this study, CsupFAR2 is confirmed to be involved in sex pheromone biosynthesis of C. suppressalis. Previous studies have demonstrated the interplay between the abundance of the pheromone fatty acyl precursors and pgFARs in shaping the pheromone composition [41]. Although CsupFAR2 shows activity on C16–C18 fatty acid substrates, it is interesting that CsupFAR2 shows high selectivity for the Z11-16:acid when both Z11-16:acid and Z9-16:acid are present as substrates (Fig 6d, i). Considering the substrate preference of CsupFAR2, the expression of CsupYPAQ and CsupKPSE in combination with CsupFAR2 suggested that the FADs and FARs are both engaged and of importance in shaping the ratio of pheromone components in C. suppressalis.
Fatty alcohols may be pheromone components for many moth species but often fatty alcohols are transformed to aldehyde pheromone components [27,28,29]. Since the 1980s, studies about moth aldehyde pheromone biosynthesis are diverging and non-conclusive [27, 42,43,44,45]. The enzyme that produces pheromone aldehydes, and thus the gene encoding it, has not been characterized. We heterologously expressed six fatty alcohol oxidases (FAO)/dehydrogenases (ADH)-like genes in N. benthamiana leaves but did not get any conclusive results indicating oxidation by any of the candidates. We observed a small amount of Z11-16:Ald in the leaf extracts when Z11-16:OH was produced in large amounts, but this occurred also without heterologous expression of FAO/ADH candidates (Fig. 7). The aldehyde production, in this case, may thus be due to an endogenous FAO/ADH activity from N. benthamiana. At the same time, it is interesting to note that all infiltrations of N. benthamiana leaves with functional FAD and FAR genes, with or without the acetyltransferase, made the plant release a remarkable amount of Z11-16:Ald as volatiles (Fig. 8c, d). The same infiltrated leaves contained very small amounts of Z11-16:Ald in the leaf extracts (Fig. 8e). This result provides evidence from a plant to support the hypothesis of Foster and Anderson (2019) [44] that in moth females the alcohols and aldehydes are produced and/or stored in different compartments of the gland. In H. virescens, they found the aldehydes mainly on the cuticle of the gland whereas the alcohols were found inside the gland beneath the cuticle. In our study, Z11-16:Ald was abundant in the volatiles and less so in the leaf extracts, which implies that the alcohol component Z11-16:OH produced by pgFAR might have been converted to aldehyde when or after it went through the leaf wax cuticle (Fig. 8a).
Acetate pheromone compounds are postulated to be produced by an acetyltransferase from its alcohol precursor. Until now, the genes encoding moth acetyltransferases involved in pheromone biosynthesis have escaped identification. Previously, a plant-derived gene EaDAcT that produces 3-acetyl-1,2-diacyl-sn-glycerols (acetyl-TAG) by the acetylation of diacylglycerol and acetyl-CoA [46] was heterologously expressed in N. benthamiana for the production of C14–C16 acetate pheromone compounds with significant but poor activity [5]. Recently, Mateos-Fernández et al. [47] also expressed EaDAcT with Atr∆11 and HarFAR in N. benthamiana, which resulted in the production and release of Z11-16:OH and Z11-16:OAc. In the present study, the ATF1 from S. cerevisiae which is naturally responsible for the synthesis of volatile esters that contribute to the fruity aroma of fermented alcoholic beverages [48] was co-expressed with moth FADs and a FAR in N. benthamiana. It resulted in the production of a high amount of acetate (Fig. 8e) compared to the previous study with EaDAcT [5]. This result is consistent with activity differences in the production of acetate pheromone compounds when ATF1 and EaDAcT were expressed in S. cerevisiae [30].
Efficient transient expression of insect genes in plant leaves for pheromone production was demonstrated by Ding et al. [5]. Plant-produced pheromones or immediate precursors have been confirmed to perform favorably compared to conventionally chemistry-produced pheromones for trapping male moths [5, 49] and are ready to be introduced for pest management [50]. “Pheromone plants” engineered for the production of insect pheromones may be applied for pest control following at least two different strategies. One strategy is the production of pheromones/pheromone precursors in the plants, isolation, and down-stream processing of the target compounds when the plant tissue has been harvested, for subsequent use of the active ingredient for moth trapping and mating disruption. The other strategy involves developing genetically modified plants capable of releasing pheromone compounds into the environment, and the pheromone-producing plant serves as a dispenser and may directly influence the behavior of insects in the environment.
Optimization of plants as producers and actual dispensers of insect pheromones requires different measures compared to engineering of plants for efficient storage of the same compounds or their precursors. For acetate release, not only a functional acetyltransferase has to be present to produce acetate, but the product has to be released, which makes it important to understand the secretion mechanism of plant volatiles. In a large number of plants, trichomes (tiny specialized hair structures for secondary metabolite production) play a prominent role for release of volatiles. Biosynthesis of plant diterpenes occurs in trichome heads, where secretory vesicles and cells are located [51,52,53]. We found that the plant carrying ATF1 controlled by pCYP71D16, a trichome-specific promoter cloned from N. tabacum, released 3- to 10-fold higher amounts of Z11-16:OAc and also much higher amounts of Z11-16:Ald and Z11-16:OH compared to when expressed from p35S (Fig. 8d). This suggests that the release of pheromone compounds benefits from the expression of the pheromone biosynthetic genes in the trichomes. More pheromones might be released along with the native plant volatiles when they are produced in trichomes.
Following several proof-of-concept studies demonstrating the possibility of producing moth pheromones in plants and in microbial cell factories [50], we now show that plants can be engineered to actually release the moth pheromone compounds. This paves the way for the stable transformation of plants required to eventually make use of moth pheromone-releasing plants in different IPM strategies. Development to successful application will depend on the outcome of challenging and exciting behavioral and ecological studies. Sex pheromones are usually mixtures of pheromone components and the behavioral response to a blend of pheromone components depends not only on the presence of components but also on the ratio between the components and their release rates. The behavioral activity of the volatiles released by the genetically modified plants should be assayed by examining different behaviors, attraction, and repellency and mating disruption in specific moth species. Our plants produce a mixture of Z11-16:OAc, Z11-16:Ald, and Z11-16:OH and all three compounds happen to be sex pheromone components of the diamondback moth Plutella xylostella. This species would thus be a first suitable candidate for behavioral experiments. Pheromone release from our plants is produced by transient expression of the relevant genes and release rates (amounts) and ratios produced vary from one experiment to the other. Thus obtaining true replicates for a behavioral assay will be a challenge. Electrophysiological experiments to demonstrate the antennal activity of the plant-produced volatiles would be easier to perform but at the same time not really add any conclusive information on “the behavioral activity” of the plants, which is what is of ultimate interest.
Conclusions
We successfully made N. benthamiana release the moth pheromone compounds Z11-16:Ald, Z11-16:OH, and Z11-16:OAc, by transient expression of the functionally characterized C. suppressalis ∆11 desaturase CsupYPAQ, the fatty acyl reductase CsupFAR2, and the yeast acetyltransferase gene ATF1. Beyond the aims of our current study, this paves the way for stable transformation and further studies of such pheromone-releasing plants in IPM strategies.
Methods
Sequences and phylogenetic analysis
The open reading frames (ORFs) of the FAD-like genes CsupYPAQ (GenBank accession number: MN453822) [54] and CsupKPSE (MN453823) [55] and the FAR-like gene CsupFAR2 (MN453825) [56] were obtained from the C. suppressalis pheromone gland transcriptome sequences [35]. To identify FAO/ADH-like genes, we retrieved from NCBI sequences corresponding to genes encoding enzymes with similar functions (Table 1). These sequences were then used to query the C. suppressalis transcriptome using BLAST [57]. This resulted in the identification of the following gene candidates: CsupFAO_15570, CsupFAO_9572, CsupADH_10975, CsupADH_14583, and CsupADH_17286 (Additional file 1: Table S2). The ORF of the ADH-like gene HzeaADH7 was obtained from the transcriptome sequences reported by Dou et al. [36, 58]. CsupYPAQ, CsupKPSE, and CsupFAR2 correspond to CsupDes4, CsupDes1, and CsupFAR2 in [35]. The AtrΔ11 (JX964774) [59] and CpuFatB1 (KC675176) [60] were amplified from entry clones [5]. The promoter pCYP71D16 was cloned from Nicotiana tabacum genome as described in Wang et al. [61].
The phylogenies of the FAD and FAR sequences were constructed using C. suppressalis FADs and FARs with the functionally characterized sequences retrieved from the GenBank from National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/). The Neighbor-Joining tree was constructed using MEGA version 5.0 [62]. The bootstrap consensus tree inferred from 500 replicates was taken to represent the evolutionary history of the analysis of the genes.
Cloning of gene candidates for functional assay
All the gene candidates were synthesized by Invitrogen, Life Technologies. PCR amplification of each gene candidate was performed using the synthesized sequence as the template with a pair of specific primers (Additional file 1: Table S3) with attB1 and attB2 sites incorporated on a Veriti Thermo Cycler using Phusion Flash High-Fidelity PCR Master Mix (Thermo Scientific ™). Cycling parameters were as follows: an initial denaturing step at 98 °C for 30 s, 38 cycles at 98 °C for 5 s, 55 °C for 10 s, 72 °C for 50 s, followed by a final extension step at 72 °C for 10 min. The PCR products were subjected to agarose gel electrophoresis and purified using the GeneJET Gel Extraction Kit (Thermo Scientific™). Then the ORFs were cloned into the pDONR221 vector in presence of BP clonase (Life Technologies) to generate the entry clone. After the entry clone for each ORF was confirmed by sequencing with M13+ and M13- primers, it was cloned into either the yeast expression vectors pYEX-CHT or pYES52 or the plant expression vector pXZP393 by LR reaction (Invitrogen). The resulting expression clones were analyzed by sequencing.
Yeast heterologous expression
The experimental workflow is shown in Additional file 1: Fig. S1a. The expression clones containing CsupYPAQ and CsupKPSE were introduced into the double deficient ole1/elo1 strain (MATa elo1::HIS3 ole1::LEU2 ade2 his3 leu2 ura3) of the yeast S.c., while the expression clones containing CsupFAR2, CsupYPAQ-CsupFAR2, pYEX-CHT empty vector, and pYES52 empty vector were introduced into the INVSc strain (MATa HIS3 LEU2 trp1-289 ura3-52), using the S.c. easy yeast transformation kit (Life technologies). For selection of uracil prototrophs, the transformed yeast was allowed to grow on SC plate containing 0.7% of YNB (w/o aa, with ammonium sulfate) and a complete drop-out medium lacking uracil (Formedium™ LTD, Norwich, England), 2% of glucose, 1% of tergitol (type Nonidet NP-40, Sigma-Aldrich Sweden AB, Stockholm, Sweden), 0.01% of adenine (Sigma). For cultivation of the double deficient ole1/elo1 strain the medium was supplemented with 0.5 mM methyl (Z)-10-heptadecanoate (Z10-17:Me) (Sigma) as an extra fatty acid source.
After 4 days (2 days for INVSc strain) incubation at 30 °C, three to five individual colonies were picked independently to inoculate 2 mL selective medium and then grown at 30 °C and 300 r.p.m for 48 h. Yeast cultures were diluted to an OD600 of 0.4 in 5 mL fresh selective medium containing 0.5 mM CuSO4 (for pYEX-CHT vector) or 10% galactose (for pYES52 vector) for induction. Then the yeast cells (medium was also harvested for CsupFAR2 and CsupYPAQ-CsupFAR2 yeast line) were harvested after 48 h incubation in a shaking incubator at 30 °C for fatty acid or alcohol analysis.
Nicotiana benthamiana material and growth condition
The wild type N. benthamiana plants for the Agrobacterium infiltration were grown in the greenhouse under 16 h/8 h light/dark conditions. The greenhouse's growth temperature and relative humidity were set at 24°C/18°C day/night and 40% RH.
Agrobacterium infiltration of Nicotiana benthamiana
The experimental workflow is shown in Additional file 1: Fig. S1b. The genes transformed for pheromone production in plants were generally controlled by the Cauliflower mosaic virus 35S promoter (p35S) and Octopine Synthase gene terminator (tOCS) but ATF1 was in one version also controlled by the tobacco trichome-specific promoter pCYP71D16. The expression clones containing CsupYPAQ, CsupKPSE, Atr∆11, CpuFatB1, ATF1, CsupFAR2, CsupFAO_15570, CsupFAO_9572, CsupADH_10975, CsupADH_14583, CsupADH_17286 and HzeaADH7 were introduced into Agrobacterium tumefaciens GV3101 strain (MP90RK) by electroporation (1700 V mm−1, 5 ms, Eppendorf 2510). A viral silencing suppressor protein P19 was introduced into GV3101 strain as well in order to inhibit the host cells’ transgene silencing apparatus and extend transgene expression over a longer period of time with a higher degree of expression [63].
The Agrobacterium infiltration of N. benthamiana started with cultivation of 10 mL of Agrobacterium that contained individual gene constructs at 30 °C with LB medium supplemented with appropriate antibiotics overnight in a 300 r.p.m. incubator. Then 100 μM acetosyringone was added to the culture and grown for an additional 2-3 h to induce virulence genes encoded by Agrobacterium genome. Subsequently, the Agrobacterium were spun down at 4200 g for 5 min at room temperature and resuspended in infiltration buffer (5 mM MgCl2, 5 mM 4-morpholineethanesulfonic acid, 100 μM acetosyringone, pH 5.7). Then the optical density under 600 nm wavelength (OD600nm) of each Agrobacterium culture was measured to adjust the final concentration of each culture to OD600nm=0.2, in a total volume of 20 mL infiltration buffer as described before [5].
Afterwards, each final mixture of Agrobacterium cells was drawn up into a 1-mL syringe without needle and infiltrated into the underside of a suitable 4-week-old N. benthamiana leaf, with a gentle squeeze on the plunger and modest pressure on the leaf using a finger. By this, the Agrobacterium solution was forced into the mesophyll spaces wetting the leaf. Five leaves of similar age from three randomly selected 4-week-old individual N. benthamiana plants were infiltrated. Then, plants were maintained in the growth chamber for 4 days with sufficient watering. One healthy infiltrated leaf from each plant was analyzed. Two times of independent gene functional assay experiments were performed in this study, each time including three to five replicates.
Lipid extraction and preparation
For yeast lipid analysis, all the cells and media (for alcohol analysis) were extracted by 1 mL of methanol/chloroform (2:1, v/v), and then 1 mL of water was added to produce a biphasic mixture, which was then vortexed vigorously and centrifuged at 2000 r.p.m for 2 min. Then ca. 330 μL of the chloroform phase containing the total lipids and pheromone compounds was transferred to a new glass vial, followed by evaporation to dryness under a gentle flow of nitrogen. The residues were used for fatty acid and pheromone analysis. For fatty acid analysis, 1 mL of 2% sulfuric acid in methanol was added, and the sample was incubated 1 h at 90 °C for methanolysis. Subsequently, 1 mL water and 1 mL heptane were added and the mixture was vortexed energetically. Finally, ca. 1 mL of heptane phase containing the fatty acids in the form of corresponding methyl esters was transferred to a new glass vial for GC/MS analysis. For pheromone compounds analysis, 200 μL heptane was added after the evaporation to dryness and after vortex vigorously the sample was then transferred to a new GC/MS analytical glass vial.
For leaf lipid analysis, ca. 100 mg of fresh leaf tissue from each sample was collected, and 3.12 μg internal standard methyl nonadecanoate (19:Me) or 1.8 μg internal standard (Z)-8-tridecenyl acetate (Z8-13:OAc) was added per gram fresh leaf to the samples for fatty acid and pheromone compound analysis respectively, following the same protocol as described above.
Sampling volatile compounds in static plant headspace
The setup for plant static headspace volatile collection is shown in Fig. 8b. After 3–5 days of infiltration, the plant was used for the collection of volatiles. The infiltrated plant leaf was enclosed in a glass funnel and covered by a transparent and odorless oven bag, and a solid-phase microextraction (SPME) fiber (65 μm film thickness, polydimethylsiloxane/divinylbenzene (PDMS/DVB), Supelco, Bellefonte, PA) was inserted from the stem of the funnel. The volatiles were collected for 24 h before analysis by gas chromatography/mass spectrometry (GC/MS). The funnel was washed with ethanol and acetone between collections. The collections for each treatment comprised six biological replicates in total. Synthetic Z11-16:OH was used as an external standard to quantify the target compounds.
Gas chromatography/mass spectrometry (GC/MS)
Yeast and plant-leaf samples were analyzed using an Agilent 5975 mass selective detector coupled to an Agilent 6890 series gas chromatograph equipped with a polar column (HP-INNOWax, 30 m × 0.25 mm, 0.25 μm film thickness) or using an Agilent 5975C mass selective detector coupled to an Agilent 7890A series gas chromatograph equipped with a non-polar column (HP-5MS, 30 m × 0.25 mm, 0.25 μm film thickness). The compounds were identified based on mass spectra and retention times on two columns being identical to synthetic standards. Helium was used as carrier gas (average velocity 33 cm/s). The injector was configured in splitless mode at 250 °C. The oven temperature was set at 80 °C for 1 min, then increased to 230 °C at a rate of 10°C/min and held for 10 min.
DMDS derivatization was performed to determine the position of double bonds in target compounds, according to Dunkelblum et al. [64]. The DMDS-adducts were analyzed by GC/MS equipped with the non-polar column (HP-5MS) under the following oven temperature program: 80 °C for 2 min, then increased at a rate of 15 °C/min to 140 °C, and then increased at a rate of 5 °C /min to 260°C, and held for 3 min.
Chemicals
Fatty acids references and synthetic pheromone compounds of various origins were available from our laboratory collection.
Statistical analysis
Data were processed by Prism version 8.0.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request. Sequences of the fatty acyl desaturases CsupYPAQ, CsupKPSE and reductase CsupFAR2 have been deposited in GenBank (accession numbers MN453822–MN453823, MN453825).
Abbreviations
- 16:OH:
-
palmityl alcohol
- Z9-16:OH:
-
(Z)-9-hexadecenol
- Z11-16:OH:
-
(Z)-11-hexadecenol
- Z13-18:OH:
-
(Z)-13-octadecenol
- Z9,Z12,Z15-18:OH:
-
linolenyl alcohol
- 14:Me:
-
Methyl myristate
- 16:Me:
-
Methyl palmitate
- 18:Me:
-
Methyl stearate
- Z9-14:Me:
-
Methyl (Z)-9-tetradecenoate
- Z9-16:Me:
-
Methyl (Z)-9-hexadecanoate
- Z11-16:Me:
-
Methyl (Z)-11-hexadecenoate
- Z9-18:Me:
-
Methyl (Z)-9-octadecenoate
- 7,10,13-16:Me:
-
Methyl hexadecatrienoate
- Z9,Z12-18:Me:
-
Methyl linoleate
- Z9,Z12,Z15-18:Me:
-
Methyl linolenate
- 16:acid:
-
palmitic acid
- Z9-16:acid:
-
(Z)-9-hexadecenoic acid
- Z9-18:acid:
-
oleic acid
- Z11-16:acid:
-
(Z)-11-hexadecenoic acid
- Z13-18:acid:
-
(Z)-13-octadecenoic acid
- Z7,Z10,Z13-16:acid:
-
roughanic acid
- Z9,Z12-18:acid:
-
linoleic acid
- Z9,Z12,Z15-18:acid:
-
α-linolenic acid
- IPM:
-
Integrated pest management
- FAD:
-
Fatty acyl desaturase
- FAR:
-
Fatty acyl reductase
- pgFAR:
-
Pheromone-gland specific fatty acyl reductase
- ATF:
-
Acetyltransferase
- FAO:
-
Fatty alcohol oxidase
- ADH:
-
Alcohol dehydrogenase
- Z11-16:OAc:
-
(Z)-11-hexadecenyl acetate
- Z11-16:Ald:
-
(Z)-11-hexadecenal
- Z13-18:Ald:
-
(Z)-13-octadecenal
- Z8-13:OAc:
-
(Z)-8-tridecenyl acetate
- Z9-16:Ald:
-
(Z)-9-hexadecenal
- Z10-17:Me:
-
Methyl (Z)-10-heptadecenoate
- 19:Me:
-
Methyl nonadecanoate
- acetyl-TAG:
-
3-acetyl-1,2-diacyl-sn-glycerols
- GC/MS:
-
Gas chromatography/mass spectrometry
- SPME:
-
Solid-phase microextraction
- PDMS/DVB:
-
polydimethylsiloxane/divinylbenzene
References
Trematerra P. Integrated pest management of stored-product insects: practical utilization of pheromones. Anzeiger für Schädlingskunde, Pflanzenschutz, Umweltschutz. 1997;70:41–4. https://doi.org/10.1007/BF01996919.
Witzgall P, Kirsch P, Cork A. Sex pheromones and their impact on pest management. J Chem Ecol. 2010;36:80–100. https://doi.org/10.1007/s10886-009-9737-y.
Mori K. New syntheses of 1, 7-dimethylnonyl propanoate, the western corn rootworm pheromone, in four different ways via cross metathesis, alkylation and coupling reactions. Biosci Biotechnol Biochem. 2010;74:595–600. https://doi.org/10.1271/bbb.90805.
Hagström ÅK, Wang HL, Liénard MA, Lassance JM, Johansson T, Löfstedt C. A moth pheromone brewery: production of (Z)-11-hexadecenol by heterologous co-expression of two biosynthetic genes from a noctuid moth in a yeast cell factory. Microb Cell Factories. 2013;12:125. https://doi.org/10.1186/1475-2859-12-125.
Ding BJ, Hofvander P, Wang HL, Durrett TP, Stymne S, Löfstedt C. A plant factory for moth pheromone production. Nat Commun. 2014;5:3353. https://doi.org/10.1038/ncomms4353.
Xia YH, Ding BJ, Wang HL, Hofvander P, Jarl-Sunesson C, Löfstedt C. Production of moth sex pheromone precursors in Nicotiana spp.: a worthwhile new approach to pest control. J Pest Sci. 2020;93:1333–46. https://doi.org/10.1007/s10340-020-01250-6.
Holkenbrink C, Ding BJ, Wang HL, Dam MI, Petkevicius K, Kildegaard KR, et al. Production of moth sex pheromones for pest control by yeast fermentation. Metab Eng. 2020;62:312–21. https://doi.org/10.1016/j.ymben.2020.10.001.
Löfstedt C, Wahlberg N, Millar JG. Evolutionary patterns of pheromone diversity in Lepidoptera. In: Allison JD, Cardé RT, editors. Pheromone communication in moths: evolution. Behavior and application. Berkeley: University of California Press; 2016. p. 43–82. https://doi.org/10.1525/9780520964433-005.
Ding BJ, Liénard MA, Wang HL, Zhao CH, Löfstedt C. Terminal fatty-acyl-CoA desaturase involved in sex pheromone biosynthesis in the winter moth (Operophtera brumata). Insect Biochem Mol Biol. 2011;41:715–22. https://doi.org/10.1016/j.ibmb.2011.05.003.
Hao G, Liu W, O’Connor M, Roelofs WL. Acyl-CoA Z9-and Z10-desaturase genes from a New Zealand leafroller moth species, Planotortrix octo. Insect Biochem Mol Biol. 2002;32:961–6. https://doi.org/10.1016/S0965-1748(01)00176-X.
Hao G, O'Connor M, Liu W, Roelofs WL. Characterization of Z/E11- and Z9-desaturases from the obliquebanded leafroller moth, Choristoneura rosaceana. J Insect Sci. 2002;2:26. https://doi.org/10.1673/031.002.2601.
Knipple DC, Rosenfield CL, Miller SJ, Liu W, Tang J, Ma PW, et al. Cloning and functional expression of a cDNA encoding a pheromone gland-specific acyl-CoA Δ11-desaturase of the cabbage looper moth, Trichoplusia ni. Proc Natl Acad Sci. 1998;95:15287–92. https://doi.org/10.1073/pnas.95.26.15287.
Knipple DC, Rosenfield CL, Nielsen R, You KM, Jeong SE. Evolution of the integral membrane desaturase gene family in moths and flies. Genetics. 2002;162:1737–52. https://doi.org/10.1093/genetics/162.4.1737.
Liu W, Jiao H, Murray NC, O'Connor M, Roelofs WL. Gene characterized for membrane desaturase that produces (E)-11 isomers of mono-and diunsaturated fatty acids. Proc Natl Acad Sci. 2002;99:620–4. https://doi.org/10.1073/pnas.221601498.
Liu W, Jiao H, O’Connor M, Roelofs WL. Moth desaturase characterized that produces both Z and E isomers of Δ11-tetradecenoic acids. Insect Biochem Mol Biol. 2002;32:1489–95. https://doi.org/10.1016/S0965-1748(02)00069-3.
Liu W, Rooney AP, Xue B, Roelofs WL. Desaturases from the spotted fireworm moth (Choristoneura parallela) shed light on the evolutionary origins of novel moth sex pheromone desaturases. Gene. 2004;342:303–11. https://doi.org/10.1016/j.gene.2004.08.017.
Rodríguez S, Hao G, Liu WT, Piña B, Rooney AP, Camps F, et al. Expression and evolution of Δ 9 and Δ 11 desaturase genes in the moth Spodoptera littoralis. Insect Biochem Mol Biol. 2004;34:1315–28. https://doi.org/10.1016/j.ibmb.2004.09.003.
Roelofs WL, Liu W, Hao G, Jiao H, Rooney AP, Linn CE. Evolution of moth sex pheromones via ancestral genes. Proc Natl Acad Sci U S A. 2002;99:13621–6. https://doi.org/10.1073/pnas.152445399.
Rosenfield CL, You KM, Marsella-Herrick P, Roelofs WL, Knipple DC. Structural and functional conservation and divergence among acyl-CoA desaturases of two noctuid species, the corn earworm, Helicoverpa zea, and the cabbage looper, Trichoplusia ni. Insect Biochem Mol Biol. 2001;31:949–64. https://doi.org/10.1016/S0965-1748(01)00043-1.
Serra M, Piña B, Abad JL, Camps F, Fabriàs GA. multifunctional desaturase involved in the biosynthesis of the processionary moth sex pheromone. Proc Natl Acad Sci. 2007;104:16444–9. https://doi.org/10.1073/pnas.0705385104.
Wang HL, Liénard MA, Zhao CH, Wang CZ, Löfstedt C. Neofunctionalization in an ancestral insect desaturase lineage led to rare Δ6 pheromone signals in the Chinese tussah silkworm. Insect Biochem Mol Biol. 2010;40:742–51. https://doi.org/10.1016/j.ibmb.2010.07.009.
Xia YH, Zhang YN, Ding BJ, Wang HL, Löfstedt C. Multi-Functional Desaturases in Two Spodoptera Moths with ∆11 and ∆12 Desaturation Activities. J Chem Ecol. 2019;45:378–87. https://doi.org/10.1007/s10886-019-01067-3.
Lassance JM, Groot AT, Liénard MA, Antony B, Borgwardt C, Andersson F, et al. Allelic variation in a fatty-acyl reductase gene causes divergence in moth sex pheromones. Nature. 2010;466:486–9. https://doi.org/10.1038/nature09058.
Moto KI, Yoshiga T, Yamamoto M, Takahashi S, Okano K, Ando T, et al. Pheromone gland-specific fatty-acyl reductase of the silkmoth, Bombyx mori. Proc Natl Acad Sci. 2003;100:9156–61. https://doi.org/10.1073/pnas.1531993100.
Antony B, Ding BJ, Moto KI, Aldosari SA, Aldawood AS. Two fatty acyl reductases involved in moth pheromone biosynthesis. Sci Rep. 2016;6:29927. https://doi.org/10.1038/srep29927.
Clinkenbeard KD, Sugiyama T, Moss J, Reed WD, Lane MD. Molecular and catalytic properties of cytosolic acetoacetyl coenzyme A thiolase from avian liver. J Biol Chem. 1973;248:2275–84. https://doi.org/10.1016/S0021-9258(19)44106-9.
Teal PEA, Tumlinson JH. Terminal steps in pheromone biosynthesis by Heliothis virescens and H. zea. J Chem Ecol. 1986;12:353–66. https://doi.org/10.1007/BF01020561.
Fang N, Teal PE, Tumlinson JH. PBAN regulation of pheromone biosynthesis in female tobacco hornworm moths, Manduca sexta (L.). Arch Insect Biochem Physiol. 1995;29:35–44. https://doi.org/10.1002/arch.940290105.
Hoskovec M, Luxová A, Svatoš A, Boland W. Biosynthesis of sex pheromones in moths: stereochemistry of fatty alcohol oxidation in Manduca sexta. Tetrahedron. 2002;58:9193–201. https://doi.org/10.1016/S0040-4020(02)01199-7.
Ding BJ, Carraher C, Löfstedt C. Sequence variation determining stereochemistry of a Δ11 desaturase active in moth sex pheromone biosynthesis. Insect Biochem Mol Biol. 2016;74:68–75. https://doi.org/10.1016/j.ibmb.2016.05.002.
Zhu ZR, Cheng J, Zuo W, Lin XW, Guo YR, Jiang YP, et al. Integrated management of rice stem borers in the Yangtze Delta, China. In: Vreysen MJB, Robinson AS, Hendrichs J, editors. Area-wide Control of Insect Pests: Springer; 2007. p. 373–82. https://doi.org/10.1007/978-1-4020-6059-5_35.
Nesbitt BF, Beevor PS, Hall DR, Lester R, Dyck VA. Identification of the female sex pheromones of the moth, Chilo suppressalis. J Insect Physiol. 1975;21:1883–6. https://doi.org/10.1016/0022-1910(75)90218-8.
Ohta K, Tztsuki S, Uchiumi K, Kurihara M, Fukami JI. Structures of sex pheromones of rice stem borer. Agric Biol Chem. 1976;40:1897–9. https://doi.org/10.1080/00021369.1976.10862323.
Tatsuki S. Sex pheromone of the rice stem borer, chilo suppressalis (Walker) (Lepidoptera: Pyralidae): the third component, Z-9-hexadecenal. Appl Entomol Zool. 1983;18:443–6. https://doi.org/10.1303/aez.18.443.
Xia YH, Zhang YN, Hou XQ, Li F, Dong SL. Large number of putative chemoreception and pheromone biosynthesis genes revealed by analyzing transcriptome from ovipositor-pheromone glands of Chilo suppressalis. Sci Rep. 2015;5:7888. https://doi.org/10.1038/srep07888.
Dou X, Liu S, Ahn SJ, Choi MY, Jurenka R. Transcriptional comparison between pheromone gland-ovipositor and tarsi in the corn earworm moth Helicoverpa zea. Comp Biochem Physiol D Genom Proteom. 2019;31:100604. https://doi.org/10.1016/j.cbd.2019.100604.
Roelofs WL, Rooney AP. Molecular genetics and evolution of pheromone biosynthesis in Lepidoptera. Proc Natl Acad Sci U S A. 2003;100:9179–84. https://doi.org/10.1073/pnas.1233767100a.
Helmkampf M, Cash E, Gadau J. Evolution of the insect desaturase gene family with an emphasis on social hymenoptera. Mol Biol Evol. 2015;32:456–71. https://doi.org/10.1093/molbev/msu315.
Lassance JM, Ding BJ, Löfstedt C. Evolution of the codling moth pheromone via an ancient gene duplication. BMC Biol. 2021;19:83. https://doi.org/10.1186/s12915-021-01001-8.
Moto K, Suzuki MG, Hull JJ, Kurata R, Takahashi S, Yamamoto M, et al. Involvement of a bifunctional fatty-acyl desaturase in the biosynthesis of the silkmoth, Bombyx mori, sex pheromone. Proc Natl Acad Sci USA. 2004;101:8631–6. https://doi.org/10.1073/pnas.0402056101.
Hagström ÅK, Liénard MA, Groot AT, Hedenström E, Löfstedt C. Semi–selective fatty acyl reductases from four heliothine moths influence the specific pheromone composition. PLoS One. 2012;7:e37230. https://doi.org/10.1371/journal.pone.0037230.
Morse D, Meighen E. Aldehyde pheromones in Lepidoptera: Evidence for an acetate ester precursor in Choristoneura fumiferana. Science. 1984;226:1434–6. https://doi.org/10.1126/science.226.4681.1434.
Morse D, Meighen E. Pheromone biosynthesis and role of functional groups in pheromone specificity. J Chem Ecol. 1986;12:335–51. https://doi.org/10.1007/BF01020560.
Foster SP, Anderson KG. Production and distribution of aldehyde and alcohol sex pheromone components in the pheromone gland of females of the moth Chloridea virescens. J Chem Ecol. 2019;45:9–17. https://doi.org/10.1007/s10886-018-1041-2.
Jurenka RA. 2 - Lepidoptera: Female sex pheromone biosynthesis and its hormonal regulation. In: Gary JB, Richard GV, editors. Insect Pheromone Biochemistry and Molecular Biology. Second Edition. Academic Press; 2021. p. 13–88. ISBN 9780128196281. https://doi.org/10.1016/B978-0-12-819628-1.00002-X.
Durrett TP, McClosky DD, Tumaney WA, Elzinga AD, Ohlrogge J, Pollard M. A distinct DGAT with sn-3 acetyltransferase activity that synthesizes unusual, reduced-viscosity oils in Euonymus and transgenic seeds. Proc Natl Acad Sci USA. 2010;107:9464–9. https://doi.org/10.1073/pnas.1001707107.
Mateos-Fernandez R, Gimenez EM, Gianoglio S, Quijano-Rubio A, Gavalda-Garcia J, Estelles L, et al. Production of Volatile Moth Sex Pheromones in Transgenic Nicotiana benthamiana Plants. Bio-Des Res. 2021, 2021:e9891082. https://doi.org/10.34133/2021/9891082.
Verstrepen KJ, Van Laere SD, Vanderhaegen BM, Derdelinckx G, Dufour JP, Pretorius IS, et al. Expression levels of the yeast alcohol acetyltransferase genes ATF1, Lg-ATF1, and ATF2 control the formation of a broad range of volatile esters. Appl Environ Microbiol. 2003;69:5228–37. https://doi.org/10.1128/AEM.69.9.5228-5237.2003.
Xia YH, Wang HL, Ding BJ, Svensson GP, Jarl-Sunesson C, Cahoon EB, et al. Green chemistry production of codlemone, the sex pheromone of the Codling Moth (Cydia pomonella), by metabolic engineering of the oilseed crop Camelina (Camelina sativa). J Chem Ecol. 2021;47:950–67. https://doi.org/10.1007/s10886-021-01316-4.
Löfstedt C, Xia YH. 3 - Biological production of insect pheromones in cell and plant factories. In: Gary JB, Richard GV, editors. Insect pheromone biochemistry and molecular biology. Second Edition. Academic Press; 2021. p. 89–121. ISBN 9780128196281. https://doi.org/10.1016/B978-0-12-819628-1.00003-1.
Kandra L, Wagner GJ. Studies of the site and mode of biosynthesis of tobacco trichome exudate components. Arch Biochem Biophys. 1988;265:425–32. https://doi.org/10.1016/0003-9861(88)90145-2.
Duke SO. Glandular trichomes-a focal point of chemical and structural interactions. Int J Plant Sci. 1994;155:617–20. https://doi.org/10.1086/297200.
Guo Z, Wagner GJ. Biosynthesis of labdenediol and sclareol in cell-free extracts from trichomes of Nicotiana glutinosa. Planta. 1995;197:627–32. https://doi.org/10.1007/BF00191570.
Xia YH, Dong SL, Zhang Y. Chilo suppressalis clone YPAQ fatty acyl CoA desaturase mRNA, complete cds. NCBI GenBank. 2019; https://www.ncbi.nlm.nih.gov/nuccore/MN453822.1.
Xia YH, Dong SL, Zhang Y. Chilo suppressalis clone KPSE fatty acyl CoA desaturase mRNA, complete cds. NCBI GenBank. 2019; https://www.ncbi.nlm.nih.gov/nuccore/MN453823.
Xia YH, Dong SL, Zhang Y. Chilo suppressalis clone FAR2 fatty acyl reductase mRNA, complete cds. NCBI GenBank. 2019; https://www.ncbi.nlm.nih.gov/nuccore/MN453825.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–10. https://doi.org/10.1016/S0022-2836(05)80360-2.
Dou X, Liu S, Ahn SJ, Choi MY, Jurenka R. RNA-Seq of Helicoverpa zea: adult female pheromone gland. NCBI SRA. 2019; https://www.ncbi.nlm.nih.gov/sra/SRX5766628%5baccn.
Ding BJ, Hofvander P, Wang HL, Durrett TP, Stymne S, Lofstedt C. Amyelois transitella delta 11 desaturase mRNA, complete cds. NCBI GenBank. 2014; https://www.ncbi.nlm.nih.gov/nuccore/JX964774.
Tjellstrom H, Strawsine M, Silva J, Cahoon EB, Ohlrogge JB. Cuphea avigera var. pulcherrima FatB type acyl-ACP thioesterase-1 (FatB1) mRNA, complete cds. NCBI GenBank. 2014; https://www.ncbi.nlm.nih.gov/nuccore/KC675176.1.
Wang E, Gan S, Wagner GJ. Isolation and characterization of the CYP71D16 trichome-specific promoter from Nicotiana tabacum L. J Exp Bot. 2002;53:1891–7. https://doi.org/10.1093/jxb/erf054.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9. https://doi.org/10.1093/molbev/msr121.
Canto T, Uhrig JF, Swanson M, Wright KM, MacFarlane SA. Translocation of Tomato bushy stunt virus P19 protein into the nucleus by ALY proteins compromises its silencing suppressor activity. J Virol. 2006;80:9064–72. https://doi.org/10.1128/JVI.00953-06.
Dunkelblum E, Tan SH, Silk PJ. Double-bond location in monounsaturated fatty acids by dimethyl disulfide derivatization and mass spectrometry: application to analysis of fatty acids in pheromone glands of four Lepidoptera. J Chem Ecol. 1985;11:265–77. https://doi.org/10.1007/BF01411414.
Acknowledgements
We thank Carin Jarl-Sunesson for helpful advice on plant cultivations and Jean-Marc Lassance for valuable discussions. We thank Erling Jirle for excellent technical support. PH recognizes support from the strategic research program Trees and Crops for the Future (TC4F).
Funding
Open access funding provided by Lund University. This work was supported by funding from the Swedish Foundation for Strategic Research (No. RBP 14-0037, Oil Crops for the Future), the European Union’s Horizon 2020 research and innovation programme (No. 760798, OLEFINE), Formas (No. 2010-857 and 2015-1336), from the Carl Trygger Foundation for Scientific Research (No. CTS 14:307 and CTS KF17:15) to CL, and the Jörgen Lindström’s Scholarship Fund and the Royal Physiographic Society in Lund to YHX. The Chinese Scholarship Council supported Yi-Han Xia’s PhD scholarship.
Author information
Authors and Affiliations
Contributions
YHX, BJD, and CL conceived the study. YHX and BJD carried out vector design and sequencing. YHX performed gene functional assays. YHX performed plant cultivation and sample analysis. HLW contributed to plant volatile collections. BJD, HLW, PH, and CL provided technical guidance and suggestions on metabolic engineering strategies. YHX drafted the manuscript with assistance from CL. All authors contributed to editing the draft. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
YHX, HLW, BJD, PH, and CL are co-inventors on patent applications including methods for production of insect pheromones in plants.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1
. Expression vectors used for functional assays in this study. Table S2. Fatty alcohol oxidase and alcohol dehydrogenase gene sequences. Table S3. Primers used in this study. Figure S1. Experimental workflow of the heterologous expression in a) yeast and b) plant.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xia, YH., Ding, BJ., Dong, SL. et al. Release of moth pheromone compounds from Nicotiana benthamiana upon transient expression of heterologous biosynthetic genes. BMC Biol 20, 80 (2022). https://doi.org/10.1186/s12915-022-01281-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12915-022-01281-8