
Abstract
Background
There are approximately 8,000 different rare diseases that affect roughly 400 million people worldwide. Many of them suffer from delayed diagnosis. Ciliopathies are rare monogenic disorders characterized by a significant phenotypic and genetic heterogeneity that raises an important challenge for clinical diagnosis. Diagnosis support systems (DSS) applied to electronic health record (EHR) data may help identify undiagnosed patients, which is of paramount importance to improve patients’ care. Our objective was to evaluate three online-accessible rare disease DSSs using phenotypes derived from EHRs for the diagnosis of ciliopathies.
Methods
Two datasets of ciliopathy cases, either proven or suspected, and two datasets of controls were used to evaluate the DSSs. Patient phenotypes were automatically extracted from their EHRs and converted to Human Phenotype Ontology terms. We tested the ability of the DSSs to diagnose cases in contrast to controls based on Orphanet ontology.
Results
A total of 79 cases and 38 controls were selected. Performances of the DSSs on ciliopathy real world data (best DSS with area under the ROC curve = 0.72) were not as good as published performances on the test set used in the DSS development phase. None of these systems obtained results which could be described as “expert-level”. Patients with multisystemic symptoms were generally easier to diagnose than patients with isolated symptoms. Diseases easily confused with ciliopathy generally affected multiple organs and had overlapping phenotypes. Four challenges need to be considered to improve the performances: to make the DSSs interoperable with EHR systems, to validate the performances in real-life settings, to deal with data quality, and to leverage methods and resources for rare and complex diseases.
Conclusion
Our study provides insights into the complexities of diagnosing highly heterogenous rare diseases and offers lessons derived from evaluation existing DSSs in real-world settings. These insights are not only beneficial for ciliopathy diagnosis but also hold relevance for the enhancement of DSS for various complex rare disorders, by guiding the development of more clinically relevant rare disease DSSs, that could support early diagnosis and finally make more patients eligible for treatment.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Background
There are approximately 8,000 rare diseases that affect about 400 million people worldwide. Most clinicians have limited knowledge about these diseases [1]. Moreover, several of them are characterized by a very high clinical and genetic heterogeneity. All these factors lead to underdiagnosis, misdiagnosis or delayed diagnosis of rare diseases. In order to accelerate the diagnosis process, which is of major importance so that patients can have access to appropriate support, personalized care and can benefit from treatments, one solution consists in automatically extracting phenotypes from patients’ electronic health records (EHRs) [2] and developing algorithms to diagnose them based on their phenotypes. It has been shown that narrative documents are mainly used by clinicians to report symptoms and comorbidities [3, 4]. This is even more important for rare diseases where patients’ clinical histories are reported by clinicians in text, as illustrated recently for Myrhe [5] and Dravet [6, 7] syndromes. More generally, recent studies showed that text reports provide much more phenotypic information [8,9,10] than structured data for models predicting diagnosis. Considering this unstructured information within text reports for diagnosis purpose is of major importance for rare diseases as early diagnosis can improve the management and progression of the disease [8]. In a recent review [11], we showed that several efforts have been made to develop diagnosis support systems (DSSs) for rare diseases, which can be categorized into three groups based on the number of targeted diseases: one specific disease, a group of diseases and the whole spectrum of rare or genetic diseases. Until 2019, almost all systems relying on phenotypes were disease recommendation systems dedicated to all rare diseases that work as follows: (1) each rare disease is described by a set of phenotype concepts that correspond to the signs and symptoms of the disease, generally encoded with the Human Phenotype Ontology (HPO) [12]; (2) Possible diagnoses of a new patient are then scored by comparing the phenotypic description of the patient to such knowledge using similarity metrics; (3) The system then returns a list of diseases ranked by the similarity score for each patient. By consequence, the use of such a system is based on the assumption that the tested patient has a rare disease, and the objective is to identify the correct one. These systems can be used in clinical practice to provide diagnostic support to non-expert clinicians in simple cases or to help domain experts select patients of interest for further investigation in complex cases. However, such systems were not designed for automated large-scale detection, and their performances in the context of complex diseases with rapidly evolving and incomplete knowledge bases are unclear. Since 2020, systems using machine learning and targeting a single disease have started to be developed [13,14,15,16,17,18,19,20]. In contrast with the disease recommendation systems, these approaches consider large scale clinical databases containing both rare and common diseases and they rely on machine learning to derive models from the data and classify patients.
Among rare diseases, ciliopathies perfectly illustrate the potential value and issues raised by patient data availability to improve diagnosis accuracy. Ciliopathies, notably those due to defects in the primary cilium, are an expanding group of severe and rare monogenic disorders with an estimated prevalence of 1/2000. So far, more than 50 ciliary disorders linked to variants in about 180 established ciliopathy-associated genes implying both phenotypic and genetic overlaps have been reported [21]. As primary cilia are ubiquitous cellular organelles, their dysfunction can lead to a large spectrum of manifestations [22] affecting mainly the kidneys, eyes, brain, liver and bone [23], among which kidney dysfunction leading to end stage kidney disease is a major cause of morbidity and mortality. Moreover, recent studies have demonstrated that renal ciliopathies are largely underdiagnosed [24]. Indeed, the rarity of the disease combined with the important phenotypic and genetic heterogeneity [25] make ciliopathies easy to confuse with other rare and common diseases and very difficult to diagnose by non-specialized clinicians. Being able to diagnose ciliopathy patients as early as possible is of major importance, so that they can benefit from appropriate support and potential future treatments. For example, a potential treatment for renal ciliopathies has been recently investigated with promising results [26]. Considering DSSs to help clinicians for the diagnosis of ciliopathies by taking advantage of all the information buried in textual reports could be a way to find undiagnosed ciliopathy patients and alleviate diagnosis wandering.
In the present study, our objective is to test existing DSSs with patient phenotypic data derived from their EHRs from an academic children’s hospital and to assess their performances for the detection of ciliopathy patients. As no system has been developed yet for ciliopathies, we focus on the generic disease recommendation systems dedicated to all rare diseases. The challenges met through our analysis will be analyzed and discussed in the “Discussion” section, with the objective to derive lessons that could be of help for the design and development of future dedicated systems for rare diseases taking advantage of large-scale clinical databases. We rely on the framework provided by the American Medical Association in 2022 [27] to address issues like population representativeness through the inclusion of cases and controls, data quality, and explicitness. Such criteria are not only beneficial for ciliopathy diagnosis but also hold relevance for the wider biomedical informatics community, aiding in the enhancement of DSS for various complex rare disorders.
Methods
Databases and data encoding
The Necker Children’s Hospital is a French reference center for rare and undiagnosed diseases that hosts the Imagine Institute, a research center specializing in genetic diseases. The clinical data warehouse developed by Necker/Imagine, named Dr Warehouse [28], contains more than 9 million documents from more than 800,000 patients from Necker hospital. Within EHRs, unstructured clinical notes are used by clinicians to describe clinical signs and detailed histories of their rare disease patients and, therefore, they provide valuable resources for diagnosis purposes [29]. The high-throughput phenotyping module of Dr. Warehouse enables the automatic extraction of all types of clinical entities from EHRs, including phenotypes and diseases based on the Unified Medical Language System (UMLS) [30].
Additionally, a database dedicated to ciliopathies, named Cilio-base, aggregates structured curated information for ciliopathy patients from different clinical departments of Necker Hospital and/or genetic departments of Imagine research Institute, including diagnoses (coded by experts from Necker/Imagine using the Orphanet nomenclature) and causal genes. More than 1800 patients with proven or suspected ciliopathy disorders are included, and 1100 of them have bi-allelic variants in one causative gene identified. 215 patients from the Cilio-base were followed at Necker Children’s Hospital, i.e., had clinical records in Dr Warehouse. We focused on these 215 patients.
Patient selection
Ciliopathies are pleiotropic diseases and causal genes remain unknown for a significant number of cases. Only half of the Cilio-base patients were completely characterized with an identified causative gene and a precise clinical diagnosis, and this proportion remained the same when considering only the 215 patients from Cilio-base with EHRs in Dr Warehouse. For this reason, two datasets of cases were considered: (1) cilio_clear, for patients with a proven ciliopathy (identified pathogenic variants and a precise diagnosis) and (2) cilio_fuzzy, for patients with a suspected ciliopathy, i.e., with clinical features compatible with a ciliopathy but without pathogenic variant identified. For both datasets, patients were randomly selected, with the additional constraint for cilio_clear to cover all ciliopathy diagnoses present in patients followed at Necker hospital.
To assess the ability of the DSSs to differentiate ciliopathy patients from other patients, we included control patients from Dr Warehouse. We did not simply take a random sample of non-ciliopathy patients from Dr Warehouse, because testing the capacity of the DSSs to differentiate ciliopathy patients from patients who are completely different from ciliopathy is pointless in real-world clinical settings. As a common morbidity across several ciliopathies is renal function deterioration, we defined control patients as patients who exhibited some overlapping phenotypes with ciliopathy patients, namely, kidney defects. We reused the set of 10,462 “other-nephropathy” patients in Dr Warehouse defined in our previous study [31], i.e., nonciliopathy patients having at least one automatically extracted UMLS phenotype concept subsumed by the term kidney diseases ([C0022658]) in their EHRs.
We selected from this collection of control patients with nephrology related signs:
-
a first control dataset (named control_random) of randomly selected patients matched on age (at the date of the most recent EHR file) and number of HPO phenotypes with selected cases.
-
a second set of patients found similar with ciliopathy patients based on the patient-patient similarity methods developed in previous studies [31, 32]: the top 30 patients from the “other-nephropathy” patients who were the most similar with ciliopathy patients were reviewed by experts, and among them a total of 11 patients who were confirmed as non-ciliopathy patients were integrated into this second dataset (named control_similar).
All patient profiles were reviewed by ciliopathy experts from Necker/Imagine (SS, MZ, KB, FP, LH, TA-B) to validate their respective categories.
For each patient, the UMLS phenotype concepts extracted from his/her EHR via Dr Warehouse were converted to HPO terms using the mapping provided by the HPO consortium (HPO format-version: 1.2; data-version: releases/2019-11-08; downloaded on 2020-02-11). Phenotypes that could not be automatically converted were discarded. Before conversion to HPO, concepts directly associated with ciliopathy diagnosis (e.g., nephronophthisis) or genes (e.g., NPHP1) were removed. To ensure that each tested patient was followed at Necker Hospital with sufficient information in his/her EHR to characterize his/her condition, we focused on patients with at least 4 HPO concepts.
Characteristics of the DSSs
Most systems dedicated to rare diseases [11] use phenotype concepts encoded with HPO [12], a patient-disease similarity-based method and return a ranked list of possible diseases. With a restriction to online accessibility and functionalities allowing the easy export of the results, three systems were considered for testing: Phenomizer [33], Genetic Diseases Diagnosis based on Phenotypes [34] (GDDP) and PubCaseFinder [35].
For Phenomizer, the authors developed a statistical model assigning p values to the resulting similarity scores between a patient and a disease, which is then used to rank the candidate diseases. Phenomizer was evaluated on a simulated cohort considering different levels of noise. GDDP proposed new methods to prioritize diseases based on semantic similarities and ontological overlap. Performance was evaluated considering the correct diagnosis rate within the top k using different ranges of cut-off value k (e.g., top 10) on simulated patients and medical records. PubCaseFinder provides a disease ranking based on disease-phenotype associations extracted from both PubMed and Orphanet using a similarity measure based on Information Content. The system was evaluated on medical records by measuring the correct diagnosis rate within the top k.
The versions of the systems and websites used for this analysis are those publicly available in June 2020. For GDDP, for each patient, we entered the HPO terms manually as free text and they were translated into HPO codes by the system. For Phenomizer, for each patient, each HPO term was manually entered and validated by using the autocomplete algorithm provided by the system. For PubCaseFinder, for each patient, we imported a file containing the HPO codes. For output, all three tools allow the automatic export of ranking diagnoses encoded in Orphanet [36] (PubCaseFinder), OMIM [37] (PubCaseFinder, GDDP), or both (Phenomizer). As PubCaseFinder independently returns Orphanet- or OMIM-encoded lists of ranked diagnoses, we tested the two systems separately, referred to as PubCaseFinder_Orpha and PubCaseFinder_OMIM, respectively, in the following sections. Default parameters were used to assess the patients.
Evaluation
We re-used patients’ EHR data to assess the ability of the disease recommendation systems to differentiate ciliopathy cases from controls. A list of ciliopathies based on Orphanet (version 2.9.1) was established with all diseases that are descendants of the following Orphanet nodes: Ciliopathy (ORPHA:363,250), Ciliopathies with major skeletal involvement (ORPHA:93,426), and Joubert syndrome and related disorders (ORPHA:140,874). This list contains 72 distinct Orphanet codes mapped to 373 distinct OMIM codes and will be referred to as CIL-ORPHA (Additional Table 1) in the following text.
The three DSSs were evaluated in previous studies considering only rare disease patients with the objective to have the correct diagnosis of each patient as highly ranked as possible (i.e., measure of the overall correct diagnosis rate within the top k). Here, the situation is different: we included some control patients, and we are only interested in the diagnosis of one rare disease, i.e., ciliopathies. Consequently, the good performance of our tested DSSs is measured by:
-
the ability to rank CIL-ORPHA diagnoses as high as possible for ciliopathy patients AND.
-
the ability to rank CIL-ORPHA diagnoses as low as possible for control patients.
We considered a classification with the ranked list provided by each DSS using the following decision criterion: given a cutoff value k, a patient was classified as positive if a CIL-ORPHA diagnosis was found within the top k and as negative otherwise.
Individual results at the patient level were then aggregated per group and DSS, and several synthetic scores were computed.
As our primary objective was to detect ciliopathy patients, we first computed the true positive rate at rank k (TPR@k), or sensitivity at rank k, defined as the proportion of ciliopathy cases classified as positive. We then assessed the specificity of the DSS by computing the false positive rate at rank k (FPR@k), defined as the proportion of controls classified as positive. We eventually plotted the receiver operating characteristic (ROC) curve, which synthesizes these two indicators.
Statistics and implementation
Associations between age, the number of HPO phenotypes and the rank of the target diagnosis were assessed with the Spearman correlation coefficient. The distributions of the rank of the target diagnosis for men and women were compared by performing the Kolmogorov-Smirnov test. All analyses were implemented using the R platform [38] with the tidyverse and ROCR packages.
Results
Datasets
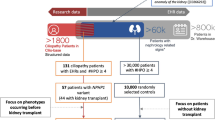
The patient selection process and the corresponding numbers of patients are summarized in Fig. 1.

Case and control datasets: pipeline and description. a Schematic overview of the patient selection process. b Description of the datasets. For each patient, age corresponds to the age at the date of the most recent EHR file. For each dataset, the three most frequent HPO terms per class of disorders using the HPO hierarchy are presented. IQR, Interquartile range; CD & CRD, Cone/cone-rod dystrophy; PPK, Palmoplantar hyperkeratosis. *CIL-ORPHA-related HPO phenotypes. †Polydipsia is classified as a nervous system disorder in the HPO but is generally associated with urine concentration defect in the context of renal ciliopathies
A total of 158 patients from Cilio-base had at least 4 HPO-encoded phenotypes in their EHRs, among whom 78 patients had a confirmed clinical and molecular diagnosis covering eleven distinct Orphanet codes of ciliopathy disorders. Among these 78 confirmed cases, we extracted a set of 60 patients covering all 11 ciliopathy Orphanet codes to build the cilio_clear data set. The distribution of diagnoses per database is provided in Additional Fig. 1. Regarding the cilio_fuzzy dataset, 30 patients were randomly selected from the 80/158 patients with suspected ciliopathies.
For the control groups, 30 patients were randomly selected as control_random matched for age and number of phenotypes with the cases. The eleven patients previously identified similar with ciliopathy patients were selected for the control_similar dataset.
An in-depth manual review by an expert was performed in order to keep only typical profiles in each class: four patients were excluded from cilio_clear, seven patients from cilio_fuzzy, and three patients from control_random (Fig. 1a), resulting in 79 ciliopathy patients and 38 controls.
The characteristics of the four datasets are shown in Fig. 1b. 86% of cases had renal impairment. 70% (55/79) of ciliopathy patients had multisystemic defects. For phenotype representation, patients in this study were associated with 1883 distinct UMLS phenotypes, and each patient was described with 64 terms on average. After conversion to the HPO, patients were associated with 792 distinct HPO terms, and each patient was described by 30 terms on average. UMLS terms that could not be mapped to HPO were either physiological characteristics that were not phenotypes in the HPO (e.g., systemic arterial pressure), or terms that were not sufficiently precise to be converted to HPO terms (e.g., cyst, fibrosis, hypertrophy). Ciliopathy patients and control_similar datasets had on average more than 50% of their HPO phenotypes associated with at least one CIL-ORPHA disease in the HPO, while patients in control_random had only 42% CIL-ORPHA-related HPO phenotypes. The most frequent HPO terms in all datasets were renal (e.g., renal insufficiency, proteinuria) and general (e.g., pain, fatigue) symptoms. Numerous neurological and skeletal disorders were found in control_similar. Disorders related to tubulointerstitial morphology were more frequent among cases than controls, and some features (e.g., polydipsia, cone-rod dystrophy) were specific to cases.
Diagnosis performances
The general performances are summarized in Fig. 2. Regardless of the DSS, the rank of the first CIL-ORPHA diagnosis was influenced neither by age nor by the number of phenotypes. We first compared the TPRs and FPRs for values of k ranging from 1 to 20 (Fig. 2a., Fig. 2b.). PubCaseFinder_OMIM obtained the best TPRs but moderate specificity, while Phenomizer had the best specificity (lowest FPRs) but very low sensitivity. The synthesis of these two indicators, as obtained by the ROC curve (Fig. 2c.), showed that PubCaseFinder_OMIM exhibited the best area under the ROC curve (AUC) (0.72), followed by Phenomizer (0.68). We compared the distributions of the ranks between cases and controls for the four DSSs (Fig. 2d.) by applying the Kolmogorov-Smirnov test. The rank of the first CIL-ORPHA diagnosis was significantly lower for cases than for controls for PubCaseFinder_OMIM and Phenomizer but not for PubCaseFinder_Orpha and GDDP.

General performances of the DSSs. a and b represent the proportion of patients classified as having ciliopathy with different cutoff k values among the cases and controls, respectively. c ROC curves and AUCs for the four DSSs. d Distribution of ranks of the first CIL-ORPHA diagnosis for the four DSSs. The red dots (resp. blue) correspond to the ranks for the two datasets of cases (resp. controls). PCF, PubCaseFinder.
We identified five situations where all DSSs failed to select a CIL-ORPHA diagnosis:
-
1.
Patients presenting with isolated symptoms were more difficult to diagnose than patients presenting with multisystemic symptoms (median rank = 12.5 vs. 5 for PubCaseFinder_OMIM).
-
2.
Situations where some key phenotypes were missing or imprecise in the EHRs (e.g., “renal insufficiency” instead of “progressive renal insufficiency”).
-
3.
Situations where some noisy phenotypes were present, and associated with incidental events (infection, diarrhea, fever, etc.).
-
4.
Situations where the phenotyping algorithm only provided generic UMLS concepts without anatomical localization, such as “cyst” (C0010709) rather than the precise phenotypes, such as “renal cyst” (C3887499).
-
5.
Some mappings between the UMLS and HPO were absent in the HPO source, e.g., for the UMLS term “Chronic kidney failure” (C0022661).
Table 1 provides a more detailed evaluation of the performances for the two DSSs with the best AUC, i.e., PubCaseFinder_OMIM and Phenomizer. Regarding cases, the TPRs were in general slightly better for cilio_clear than cilio_fuzzy with both the DSSs. PubCaseFinder_OMIM had the best TPR for k = 5, k = 10 and k = 20 for both datasets, and the performances for cilio_fuzzy were almost identical to those for cilio_clear (TPR@20 = 70% vs. 75%), showing that the system was able to detect ciliopathy even for patients with unknown pathogenic variants. In other terms, a TPR equal or higher than 70% could be achieved only with k = 20. Whatever the DSS, the TPR was always lower than 50% for k = 5 (ranging from 17 to 48%) for both cilio_clear and cilio_fuzzy. Regarding controls, the FPRs for low values of k were slightly higher for control_similar than for control_random. Phenomizer had lower FPRs for both datasets. As expected, the proportion of patients classified as ciliopathy was higher in the case datasets than in the control datasets for PubCaseFinder_OMIM and Phenomizer, which shows that, to some extent, these DSSs were able to distinguish between cases and controls, even controls exhibiting a high similarity score with cases. However, none of these systems obtained results good enough to be equivalent to “expert”.
Differential diagnoses
As PubCaseFinder_OMIM obtained the best performances, especially for low values of k, we focused on this DSS to analyze the 10 diseases most frequently ranked within the top k (with k = 5) for cases and controls (Table 2). Among them, six were found for both cases and controls. Fabry disease shares a major feature, renal dysfunction, with ciliopathies and other nephropathies. The five other differential diagnoses in common were diseases affecting multiple organs and associated with a very important number of HPO terms: these diseases belonged to the top 1% of diseases in OMIM regarding the number of HPO phenotypes. Regarding cases, the only disease present in CIL-ORPHA was Alström syndrome, which was not represented among our study population but is also associated with a very high number of HPO terms. The other differential diagnoses for ciliopathy cases were all diseases with overlapping clinical features with ciliopathies, i.e., nephropathic cystinosis, congenital disorder of glycosylation, type 1a (CDG1A), and peroxisome biogenesis disorder 1 A (Zellweger).
Discussion
In this study, we evaluated the performance of current DSSs on complex genetic diseases, and used the example of ciliopathies, a group of complex pleiotropic disorders caused by cilia dysfunction. As no dedicated DSS has been developed yet, we evaluated generic rare disease DSSs for the diagnosis of ciliopathies using all phenotypes extracted from patient EHRs. The evaluation was performed in a children’s hospital specializing in genetic diseases but also serving as a general pediatric center for the local population.
In the original paper, Phenomizer [33] outperformed other scores on a cohort of simulated patients, ranking the correct diagnosis as the first proposal in more than 75% of the cases. GDDP [34] was compared with existing methods and outperformed them on medical records (top 10 diagnosis rate = ~ 32% for GDDP vs. ~ 4% for Phenomizer and ~ 20% for BOQA [39]). PubCaseFinder [35] was compared to other tools (Phenomizer and Orphamizer) on medical records and globally reached results comparable with Phenomizer. It obtained a top 10 diagnosis rate of 57% (Phenomizer = 47%, Orphamizer = 31%). These results highlight the variability of performance depending on the dataset under study. In the present study, PubCaseFinder_OMIM obtained the best rate of true positives for k = 20 (TPR > 70%) but the TPR scores dropped to 35–48% for k = 5. Moreover, it misclassified controls with an FPR@20 higher than 40%. Phenomizer obtained the lowest rate of false positives, but its sensitivity was not high enough to identify ciliopathy patients. Overall, PubCaseFinder_OMIM exhibited the best AUC (0.72). Not surprisingly, patients with multisystemic symptoms were generally easier to diagnose than patients with isolated symptoms. This may be partly because most patients with isolated symptoms had renal impairment, which generally presents with nonspecific features [40]. To summarize, none of these systems obtained results good enough to be equivalent to “expert”. Several interrelated lessons emerged from our evaluation, and we have attempted to encapsulate the following four key lessons for the future developers and users of rare disease DSSs.
The first lesson learnt was that these DSSs should ideally be integrated into the existing healthcare ecosystem, interoperable with the EHRs and capable of leveraging EHR data. This is of major importance because unstructured clinical notes in EHRs are unique sources of clinical information for diagnosis purposes, in particular for rare diseases [29]. Analysing the EHRs of two academic health institutions, Liu et al. [41] showed that the phenotypic coverage was much higher in clinical notes (about 36% of all phenotypic concepts in HPO) than in structured data (4%), for phenotypes found in at least 10 individuals. However, they stated that the EHR were rarely explored yet to generate rare disease-phenotype associations [41]. In this study, we benefited from access to patient EHRs as well as to an automated pipeline in order to generate HPO-based phenotypic descriptions of patients. However, whatever the DSS, the data had to be entered on a patient-by-patient basis into the DSS, and one of the evaluated tools required manual input and validation of each phenotype for each patient. We claim that such a time-consuming process is a major obstacle for large external evaluation of DSSs and hinders their large-scale use in clinical practice. Moreover, the disconnection from the EHR constraints on the use of data-driven approaches. The landscape of DSS has been transformed recently in many disciplines ranging from cancer [42, 43], to COVID [44] and sepsis [45], with machine learning models developed on multi-site EHR data. However, as stressed out by Schaaf et al. in their review of clinical DSSs for rare diseases [46], machine learning has been far less developed for rare diseases than in other medical fields, whereas the availability of EHR data provides an opportunity for developing algorithms that identify patients having a high probability to have a disease from large clinical data warehouses [13,14,15,16,17,18,19,20]. Ciliopathies perfectly illustrate the potential benefits of these new approaches, as it is of paramount importance to identify patients with suspected ciliopathies before the development of irreversible lesions as a potential treatment has been recently investigated with promising results. However, most rare disease research groups do not take advantage of EHR-based data driven approaches and stay focused on traditional disease recommendation systems.
The second lesson learnt was that it is important to validate the performances of DSSs in real-life settings involving clinicians and domain experts, which can benefit from the interoperability with the EHR system by providing cases and controls and enabling evaluators to test DSS in real life conditions. The American Medical Association recently highlighted that clinicians should bring critical insights on AI applications and should be involved in shaping AI’s role [27]. Similarly, Youssef et al. highlighted that real-world studies are a mandatory step to evaluate the deployed models’ usefulness [47]. We share this vision and claim that all DSSs should have external evaluation mimicking real-life, like in the studies from Weber et al. [44] or Adam et al. [45]. Indeed, the global performance of a system may be much lower in real-world settings than the scores achieved in the test set, as shown for example for common skin diseases [48, 49] and rare cardiomyopathy [14]. This is especially true when complex pipelines are needed to extract phenotypic information from EHRS. As described in our previous work, some tools are even only evaluated on simulated patients, whereas when tested on real patients some of them obtained performances that were much lower than on simulated ones [34, 50]. For example, as highlighted in this study, Phenomizer was tested for comparison with developed tools in numerous studies and obtained results that highly depended on the dataset under study [34, 35, 51,52,53]. This is particularly important for rare diseases where the question is: among the patients having renal symptoms, who is suspected to have ciliopathy and should have genetic testing, and could potentially benefit from treatment? In our study, cases were tested in contrast to carefully selected controls. In order to mimic real situations, the controls were patients having overlapping phenotypes with ciliopathy patients but not diagnosed with ciliopathy. We think that the inclusion of such kinds of controls is of major importance, as one key difficulty with rare diseases is to differentiate them from common diseases with overlapping profiles. However, until now, most of the generic DSSs have been evaluated only on rare disease patients.
The third lesson learnt was that high-quality data is crucial to make DSSs effective. Quality issues include both EHR data quality and NLP extraction quality. As key information may be present only as text in the EHR, Garcelon et al. [54] and Schaaf et al. [55] suggested developing adequate NLP methods to extract reliable and accurate information from unstructured text. Indeed, as pointed out by recent studies [56], EHR data may include incomplete records and inaccurate information. Missing or incomplete data is an obstacle to getting a comprehensive view of a patient’s medical history, while imprecise or noisy phenotype descriptions further complicate the precise capture and representation of a patient’s profile from clinical narratives using NLP techniques [57]. For example, in our study, the Named Entity Extraction pipeline sometimes failed at coordinating the location with the phenotype into a single term: it did not extract “renal cyst” from sentences like “the renal ultrasound confirmed the presence of cysts.‘’ “Renal” and “cyst” were extracted independently and the mapping to “renal cyst” (HP:0000107) was not completed. As for imprecise EHR phenotypes, such granularity issues may lead to inappropriate disease recommendations. In a recent work [58], we proposed a hybrid method combining a dictionary-based method with deep learning to enrich the set of UMLS terms. We trained and evaluated it on a ciliopathy characterized by skeletal abnormalities, Jeune syndrome and could strongly improve the detection of phenotypes from the EHRs. We plan to adapt such a method for other ciliopathies, starting with ciliopathies with renal impairment.
The fourth lesson is related to the relevance of the algorithm. All the tested DSSs have leveraged medical ontologies such as HPO to address granularity issues, and use the associations among phenotypes, genes and diseases provided by OMIM and Orphanet to deliver a list of possible diseases. All three are fine-grained terminologies designed for rare diseases [55]. However, such knowledge bases do not reflect the state-of-the-art knowledge in many rare-diseases domains, like e.g., ciliopathies, where research is still very active. We think that transparency regarding the knowledge incorporated in the algorithm, e.g., in our case, the class/set of ciliopathy disorders defined with the algorithm, is key for the correct interpretation of the algorithm results. Moreover, complex disease subtypes exhibit important phenotypic and genetic overlap. In our study, the analysis of the top-ranked differential diagnoses suggested by PubCaseFinder_OMIM showed that several diseases, e.g., Zellweiger syndrome, were easily confused with ciliopathy because they also affect multiple organs and have overlapping phenotypes with ciliopathies. The poor performance of the DSSs suggests that the complexity of ciliopathies requires integrating more expert knowledge specific to ciliopathies into the model. Otherwise, we also observed among the top-ranked differential diagnoses an overrepresentation of diseases associated with a large number of HPO phenotypes, e.g., Rubinstein-Taybi and Williams-Beuren syndrome. This reveals the need for fine-tuning the algorithms or adequate weighting/normalization in the computation of patient-disease similarity to better match the specific characteristics of the targeted patient population. A step further, we believe that methods based on patient-patient similarity [59] should be more interesting to support early diagnosis than those based on patient-disease similarity since the real-world patient data contain a wide range of complex information. Supervised machine learning methods are another way to leverage EHR data from diagnosed patients to detect undiagnosed patients, but they are usually limited to a single diagnosis, e.g., NPHP1 pathogenic variants [60], and should be extended to broader disease coverage through multiclass models. To minimize bias that may be produced by individual variability, unsupervised methods that allow the integration of different types of information can be considered beforehand to identify clusters of ciliopathy patients [61]. A more sophisticated solution consists in using graphs to represent complex systems such as interactions between proteins, comorbidities between diseases, or healthcare knowledge [62]. In the rare disease field, they have been used to identify patients sharing similar phenotypes [63]. Adapted clustering methods have been proposed to study the graph structure and derive information from this representation [64, 65]. Such methods could help identify clusters of patients based on the graph representation. In order to make these change happen, the rare disease community will have to overcome the challenges related with the scarcity of the data [54, 66], utilize not only dedicated databases and registries but all data collected during routine healthcare processes – structured, narrative reports, genetic, etc. and favor new digital models able to combine expert knowledge and machine learning approaches.
Conclusions
Clinicians need DSSs to support diagnosis of patients that have symptoms shared by both rare and common conditions. Existing disease recommendation systems do not consider common diseases, and, although they rely on state-of-the-art semantic methods and ontologies, their performance in diagnosing highly heterogeneous rare diseases does not reach expert levels. Challenges related to interoperability, algorithm transparency, clinical validation, data quality, ontology use, and context of application have been highlighted in this study. We conclude that the effectiveness of a DSS is influenced by the model as well as by how it is integrated within the EHR system. These lessons can guide the development of more effective and clinically relevant rare disease DSSs, that could support earlier diagnosis of rare disease patients and offer new perspectives in patient management.
Availability of data and materials
The clinical datasets from this study are not publicly available, as institutional officials expressed concern about the inability to guarantee anonymity. Aggregate data and datasets containing coarse-grained phenotypes are available upon request to the corresponding author.
References
RARE Disease Facts. Global Genes. https://globalgenes.org/rare-disease-facts/. Cited 2022 Jul 8.
Colbaugh R, Glass K, Rudolf C. Tremblay Volv Global, Lausanne, Switzerland M. Learning to identify rare disease patients from electronic health records. AMIA Annu Symp Proc. 2018;2018:340–7.
Neuraz A, Lerner I, Digan W, Paris N, Tsopra R, Rogier A, et al. Natural language processing for rapid response to emergent diseases: case study of calcium channel blockers and hypertension in the COVID-19 pandemic. J Med Internet Res. 2020;22(8):e20773.
Escudié JB, Rance B, Malamut G, Khater S, Burgun A, Cellier C, et al. A novel data-driven workflow combining literature and electronic health records to estimate comorbidities burden for a specific disease: a case study on autoimmune comorbidities in patients with celiac disease. BMC Med Inf Decis Mak. 2017;17:140.
Yang DD, Rio M, Michot C, Boddaert N, Yacoub W, Garcelon N, et al. Natural history of Myhre syndrome. Orphanet J Rare Dis. 2022;17(1):304.
Lo Barco T, Kuchenbuch M, Garcelon N, Neuraz A, Nabbout R. Improving early diagnosis of rare diseases using Natural Language Processing in unstructured medical records: an illustration from Dravet syndrome. Orphanet J Rare Dis. 2021;16(1):309.
Lo Barco T, Garcelon N, Neuraz A, Nabbout R. Natural history of rare diseases using natural language processing of narrative unstructured electronic health records: The example of Dravet syndrome. Epilepsia. 2023. https://pubmed.ncbi.nlm.nih.gov/38065926/. Cited 2024 Jan 4.
Zanello G, Chan CH, Pearce DA. Recommendations from the IRDiRC Working group on methodologies to assess the impact of diagnoses and therapies on rare disease patients. Orphanet J Rare Dis. 2022;17:181.
Zhou S, Wang N, Wang L, Liu H, Zhang R. CancerBERT: a cancer domain-specific language model for extracting breast cancer phenotypes from electronic health records. J Am Med Inf Assoc. 2022:1208-16.
Kohane IS, Aronow BJ, Avillach P, Beaulieu-Jones BK, Bellazzi R, Bradford RL, et al. What every reader should know about studies using Electronic Health Record Data but May be afraid to ask. J Med Internet Res. 2021;23(3):e22219.
Faviez C, Chen X, Garcelon N, Neuraz A, Knebelmann B, Salomon R, et al. Diagnosis support systems for rare diseases: a scoping review. Orphanet J Rare Dis. 2020;15(1):94.
Robinson PN, Köhler S, Bauer S, Seelow D, Horn D, Mundlos S. The human phenotype ontology: a tool for annotating and analyzing human hereditary disease. Am J Hum Genet. 2008;83(5):610–5.
Movaghar A, Page D, Brilliant M, Mailick M. Advancing artificial intelligence-assisted pre-screening for fragile X syndrome. BMC Med Inf Decis Mak. 2022;22(1):152.
Huda A, Castaño A, Niyogi A, Schumacher J, Stewart M, Bruno M, et al. A machine learning model for identifying patients at risk for wild-type transthyretin amyloid cardiomyopathy. Nat Commun. 2021;12(1):2725.
Willis C, Watanabe AH, Hughes J, Nolen K, O’Meara J, Schepart A, et al. Applying diagnosis support systems in electronic health records to identify wild-type transthyretin amyloid cardiomyopathy risk. Future Cardiol. 2022;18(5):367–76.
Jefferies JL, Spencer AK, Lau HA, Nelson MW, Giuliano JD, Zabinski JW, et al. A new approach to identifying patients with elevated risk for fabry disease using a machine learning algorithm. Orphanet J Rare Dis. 2021;16(1):518.
Rider NL, Cahill G, Motazedi T, Wei L, Kurian A, Noroski LM, et al. PI Prob: a risk prediction and clinical guidance system for evaluating patients with recurrent infections. PLoS ONE. 2021;16(2):e0237285.
García-García E, González-Romero GM, Martín-Pérez EM, Zapata Cornejo E, de D, Escobar-Aguilar G. Cárdenas Bonnet MF. Real-world data and machine learning to predict cardiac amyloidosis. Int J Environ Res Public Health. 2021;18(3):908.
Doyle OM, van der Laan R, Obradovic M, McMahon P, Daniels F, Pitcher A, et al. Identification of potentially undiagnosed patients with nontuberculous mycobacterial lung disease using machine learning applied to primary care data in the UK. Eur Respir J. 2020;56(4):2000045.
Cohen AM, Chamberlin S, Deloughery T, Nguyen M, Bedrick S, Meninger S, et al. Detecting rare diseases in electronic health records using machine learning and knowledge engineering: case study of acute hepatic porphyria. PLoS ONE. 2020;15(7):e0235574.
Reiter JF, Leroux MR. Genes and molecular pathways underpinning ciliopathies. Nat Rev Mol Cell Biol. 2017;18(9):533–47.
Powles-Glover N. Cilia and ciliopathies: Classic examples linking phenotype and genotype—An overview. Reprod Toxicol. 2014;48:98–105.
McConnachie DJ. Ciliopathies and the Kidney: A Review. Am J Kidney Dis. 2021;77:10.
Snoek R, van Setten J, Keating BJ, Israni AK, Jacobson PA, Oetting WS, et al. NPHP1 (Nephrocystin-1) gene deletions cause adult-onset ESRD. J Am Soc Nephrol. 2018;29(6):1772–9.
Petzold F, Billot K, Chen X, Henry C, Filhol E, Martin Y, et al. The genetic landscape and clinical spectrum of nephronophthisis and related ciliopathies. Kidney Int. 2023;104(2):378–87.
Garcia H, Serafin AS, Silbermann F, Porée E, Viau A, Mahaut C, et al. Agonists of prostaglandin E2 receptors as potential first in class treatment for nephronophthisis and related ciliopathies. Proc Natl Acad Sci U S A. 2022;119(18):e2115960119.
Crigger E, Reinbold K, Hanson C, Kao A, Blake K, Irons M. Trustworthy augmented intelligence in health care. J Med Syst. 2022;46(2):12.
Garcelon N, Neuraz A, Salomon R, Faour H, Benoit V, Delapalme A, et al. A clinician friendly data warehouse oriented toward narrative reports: Dr. Warehouse. J Biomed Inform. 2018;80:52–63.
Morley TJ, Han L, Castro VM, Morra J, Perlis RH, Cox NJ, et al. Phenotypic signatures in clinical data enable systematic identification of patients for genetic testing. Nat Med. 2021;27(6):1097–104.
Bodenreider O. The Unified Medical Language System (UMLS): integrating biomedical terminology. Nucleic Acids Res. 2004;32(Database issue):D267–270.
Chen X, Garcelon N, Neuraz A, Billot K, Lelarge M, Bonald T, et al. Phenotypic similarity for rare disease: ciliopathy diagnoses and subtyping. J Biomed Inf. 2019;100:103308.
Chen X, Faviez C, Vincent M, Garcelon N, Saunier S, Burgun A. Identification of similar patients through Medical Concept Embedding from electronic health records: a feasibility study for rare disease diagnosis. Stud Health Technol Inf. 2021;281:600–4.
Köhler S, Schulz MH, Krawitz P, Bauer S, Dölken S, Ott CE, et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009;85(4):457–64.
Chen J, Xu H, Jegga A, Zhang K, White PS, Zhang G. Novel phenotype-disease matching tool for rare genetic diseases. Genet Med. 2019;21(2):339–46.
Fujiwara T, Yamamoto Y, Kim JD, Buske O, Takagi T, PubCaseFinder:. A case-report-based, phenotype-driven differential-diagnosis system for Rare diseases. Am J Hum Genet. 2018;06(3):389–99.
[Orphanet: a European database for rare diseases]. - Abstract - Europe PMC. https://europepmc.org/abstract/med/18389888. Cited 2019 Oct 24.
Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online mendelian inheritance in man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(Database issue):D514–517.
R Core Team. R: A Language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2020. https://www.R-project.org/.
Bauer S, Köhler S, Schulz MH, Robinson PN. Bayesian ontology querying for accurate and noise-tolerant semantic searches. Bioinformatics. 2012;28(19):2502–8.
Arts HH, Knoers NVAM. Current insights into renal ciliopathies: what can genetics teach us? Pediatr Nephrol. 2013;28(6):863–74.
Liu C, Ta CN, Havrilla JM, Nestor JG, Spotnitz ME, Geneslaw AS, et al. OARD: open annotations for rare diseases and their phenotypes based on real-world data. Am J Hum Genet. 2022;109(9):1591–604.
Dembrower K, Crippa A, Colón E, Eklund M, Strand F, ScreenTrustCAD trial consortium. artificial intelligence for breast cancer detection in screening mammography in Sweden: a prospective, population-based, paired-reader, non-inferiority study. Lancet Digit Health. 2023;5(10):e703–11.
Lång K, Josefsson V, Larsson AM, Larsson S, Högberg C, Sartor H, et al. Artificial intelligence-supported screen reading versus standard double reading in the Mammography screening with Artificial Intelligence trial (MASAI): a clinical safety analysis of a randomised, controlled, non-inferiority, single-blinded, screening accuracy study. Lancet Oncol. 2023;24(8):936–44.
Weber GM, Hong C, Xia Z, Palmer NP, Avillach P, L’Yi S, et al. International comparisons of laboratory values from the 4CE collaborative to predict COVID-19 mortality. NPJ Digit Med. 2022;5(1):74.
Adams R, Henry KE, Sridharan A, Soleimani H, Zhan A, Rawat N, et al. Prospective, multi-site study of patient outcomes after implementation of the TREWS machine learning-based early warning system for sepsis. Nat Med. 2022;28(7):1455–60.
Schaaf J, Sedlmayr M, Schaefer J, Storf H. Diagnosis of Rare diseases: a scoping review of clinical decision support systems. Orphanet J Rare Dis. 2020;15(1):263.
Youssef A, Pencina M, Thakur A, Zhu T, Clifton D, Shah NH. External validation of AI models in health should be replaced with recurring local validation. Nat Med. 2023;29(11):2686–7.
Zaar O, Larson A, Polesie S, Saleh K, Tarstedt M, Olives A, et al. Evaluation of the diagnostic accuracy of an online Artificial Intelligence Application for skin disease diagnosis. Acta Derm Venereol. 2020;100(16):adv00260.
Steele L, Velazquez-Pimentel D, Thomas BR. Do AI models recognise rare, aggressive skin cancers? An assessment of a direct-to-consumer app in the diagnosis of Merkel cell carcinoma and amelanotic melanoma. J Eur Acad Dermatol Venereol. 2021;35(12):e877–9.
Zemojtel T, Köhler S, Mackenroth L, Jäger M, Hecht J, Krawitz P, et al. Effective diagnosis of genetic disease by computational phenotype analysis of the disease-associated genome. Sci Transl Med. 2014;6(252):252ra123.
Ullah MZ, Aono M, Seddiqui MH. Estimating a ranked list of human genetic diseases by associating phenotype-gene with gene-disease bipartite graphs. ACM Trans Intell Syst Technol. 2015;6(4):56.
Yang H, Robinson PN, Wang K. Phenolyzer: phenotype-based prioritization of candidate genes for human diseases. Nat Methods. 2015;12(9):841–3.
Pinol M, Alves R, Teixido I, Mateo J, Solsona F, Vilaprinyo E. Rare disease discovery: an optimized disease ranking system. IEEE Trans Ind Inf. 2017;13(3):1184–92.
Garcelon N, Burgun A, Salomon R, Neuraz A. Electronic health records for the diagnosis of rare diseases. Kidney Int. 2020;97(4):676–86.
Schaaf J, Sedlmayr M, Sedlmayr B, Storf H. User-centred development of a diagnosis support system for rare diseases. dHealth. 2022;2022:11–8.
Kim E, Rubinstein SM, Nead KT, Wojcieszynski AP, Gabriel PE, Warner JL. The evolving use of electronic health records (EHR) for research. Semin Radiat Oncol. 2019;29(4):354–61.
Sarker A. LexExp: a system for automatically expanding concept lexicons for noisy biomedical texts. Bioinformatics. 2021;37(16):2499–501.
Faviez C, Vincent M, Garcelon N, Michot C, Baujat G, Cormier-Daire V, et al. Enriching UMLS-based phenotyping of rare diseases using deep-learning: evaluation on Jeune syndrome. Stud Health Technol Inf. 2022;294:844–8.
Chen X, Faviez C, Vincent M, Briseño-Roa L, Faour H, Annereau JP et al. Patient-Patient similarity-based screening of a clinical data warehouse to support ciliopathy diagnosis. frontiers in pharmacology. 2022;13. https://www.frontiersin.org/article/https://doi.org/10.3389/fphar.2022.786710. Cited 2022 Apr 4.
Faviez C, Vincent M, Garcelon N, Boyer O, Knebelmann B, Heidet L, et al. Performance and clinical utility of a new supervised machine-learning pipeline in detecting rare ciliopathy patients based on deep phenotyping from electronic health records and semantic similarity. Orphanet J Rare Dis. 2024;19(1):55.
Chen X, Faviez C, Vincent M, Saunier S, Garcelon N, Burgun A. Improving patient similarity using different modalities of phenotypes extracted from clinical narratives. Stud Health Technol Inf. 2023;302:1037–41.
Li MM, Huang K, Zitnik M. Graph representation learning in biomedicine and healthcare. Nat Biomed Eng. 2022;6(12):1353–69.
Buphamalai P, Kokotovic T, Nagy V, Menche J. Network analysis reveals rare disease signatures across multiple levels of biological organization. Nat Commun. 2021;12(1):6306.
Hu L, Pan X, Tang Z, Luo X. A fast fuzzy clustering algorithm for Complex Networks via a generalized momentum method. IEEE Trans Fuzzy Syst. 2022;30(9):3473–85.
Yang Y, Su X, Zhao B, Li G, Hu P, Zhang J, et al. Fuzzy-based deep attributed graph clustering. IEEE Trans Fuzzy Syst. 2024;32(4):1951–64.
Decherchi S, Pedrini E, Mordenti M, Cavalli A, Sangiorgi L. Opportunities and challenges for Machine Learning in Rare diseases. Front Med (Lausanne). 2021;8:747612.
Acknowledgements
The authors acknowledge URC-CIC Paris Centre for the implementation of the study. The authors thank Isabelle Perrault, Laurence Heidet, Céline Huber, Caroline Michot, Ilyas Challet and Corinne Antignac for their contributions.
Funding
This work was supported by state funding by The French National Research Agency (ANR) under the C’IL-LICO project (ANR-17-RHUS-0002) and as part of the “Investissements d’avenir” program (ANR-19-P3IA-0001) (PRAIRIE 3IA Institute). FP is funded by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG) grant PE 3135/1–1 and project number 49366873 - MD-LEICS.
Author information
Authors and Affiliations
Contributions
AB, XC, CF supported the study design. SS, KB, FP, NG, HF, MD, TAB, VCD, JMR contributed to the data curation, annotation and classification. CF, XC performed the analyses. CF, XC, AB, SS, NG, MZ, KB, FP, SL participated to the analysis of the results and writing of the manuscript. All authors critically revised and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The C’IL-LICO project and study protocol received approval from the French National Ethics and Scientific Committee for Research, Studies and Evaluations in the Field of Health (CESREES) under the number #2201437. The data processing was approved by the French Data Protection Authority (CNIL) with a waiver of informed consent under number DR-2023-017//920398v1.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12911_2024_2538_MOESM1_ESM.xlsx
Additional file 1: CIL-ORPHA list. List of ciliopathy diseases used for the classification. The list is built based on the Orphanet hierarchy and disease names are encoded using the Orphanet nomenclature.
12911_2024_2538_MOESM2_ESM.pdf
Additional file 2: Proportions of ciliopathy diagnoses per database. Proportions of diagnoses among patients with medical and genetic diagnosis for Cilio-base (in red), Cilio-base ∩ Dr. Warehouse (in purple) and cilio_clear patients (in yellow).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Faviez, C., Chen, X., Garcelon, N. et al. Objectivizing issues in the diagnosis of complex rare diseases: lessons learned from testing existing diagnosis support systems on ciliopathies. BMC Med Inform Decis Mak 24, 134 (2024). https://doi.org/10.1186/s12911-024-02538-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12911-024-02538-8