
Abstract
Background
HVC1 consists of Coptidis Rhizoma (dried rhizome of Coptischinensis), Scutellariae Radix (root of Scutellariabaicalensis), Rhei Rhizoma (rhizome of Rheum officinale), and Pruni Cortex (cortex of Prunusyedoensis Matsum). Although the components are known to be effective in various conditions such as inflammation, hypertension, and hypercholesterolemia, there are no reports of the molecular mechanism of its hypolipidemic effects.
Methods
We investigated the hypolipidemic effect of HVC1 in low-density lipoprotein receptor-deficient (LDLR−/−) mice fed a high-cholesterol diet for 13 weeks. Mice were randomized in to 6 groups: ND (normal diet) group, HCD (high-cholesterol diet) group, and treatment groups fed HCD and treated with simvastatin (10 mg/kg, p.o.) or HVC1 (10, 50, or 250 mg/kg, p.o.).
Results
HVC1 regulated the levels of total cholesterol, triglyceride (TG), low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol in mouse serum. In addition, it regulated the transcription level of the peroxisome proliferator-activated receptors (PPARs), sterol regulatory element-binding proteins (SREBP)-2, 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase, lipoprotein lipase (LPL), apolipoprotein B (apo B), liver X receptor (LXR), and inflammatory cytokines (IL-1β, IL-6, and TNF-α). Furthermore, HVC1 activated 5′ adenosine monophosphate-activated protein kinase (AMPK).
Conclusion
Our results suggest that HVC1 might be effective in preventing high-cholesterol diet-induced hyperlipidemia by regulating the genes involved in cholesterol and lipid metabolism, and inflammatory responses.
Similar content being viewed by others


Background
Hyperlipidemia is defined as increased levels of cholesterol, cholesterol esters, phospholipids, and triglycerides in the serum [1]. About 20–25% of total daily cholesterol production occurs in the liver, which is also the organ that regulates cholesterol homeostasis. Cholesterol synthesis in the liver is responsive to external factors and hence, suppressed by an increase in dietary cholesterol. Triglycerides and cholesterol both from diet and synthesized in the liver are solubilized in lipoproteins [2]. These lipoproteins contain triglyceride lipid droplets and cholesteryl esters surrounded by polar phospholipids and proteins identified as apolipoproteins. There are several types of lipoproteins in the blood. In order of increasing density, they are chylomicrons, very-low-density lipoprotein (VLDL), low-density lipoprotein (LDL), intermediate-density lipoprotein (IDL), and high-density lipoprotein (HDL). Lower protein/lipid ratios yield less dense lipoproteins. The cholesterol within different lipoproteins is identical although some is carried as “free” alcohol and others as fatty acyl esters, known also as cholesterol esters. Low-density lipoprotein receptors (LDLR) regulate cholesterol-rich IDL and LDL in serum; therefore, LDLR deficiency is related to elevated cholesterol levels, particularly IDL/LDL cholesterol [3]. LDLR−/− mice demonstrate a human-like lipoprotein profile characterized by elevated cholesterol levels [4].
Peroxisome proliferator-activated receptors (PPARs) family is nuclear receptors superfamily that regulates in energy homeostasis and metabolic function such as β-oxidation of fatty acids, cholesterol metabolism [5]. The PPAR subfamily is composed of three members including PPAR-α, PPAR-δ, and PPAR-γ [6]. PPAR-α is known to regulation of genes involved in fatty acid oxidation, gluconeogenesis, cholesterol catabolism, and lipoprotein metabolism [7, 8]. Ligands for PPAR-α reduce triglycerides and LDL-cholesterol, and increase HDL-cholesterol and cholesterol efflux by inducing the expression of LXR [9, 10]. PPAR-δ regulates the expression of oxidative enzymes, and metabolic activity in the skeletal muscles, liver, and heart [11]. In addition, PPAR-δ may improve blood lipids and lipoproteins in atherosclerotic condition [12]. PPAR-δ agonist suppressed atherosclerotic lesion by improving the serum lipoprotein profiles [13]. Some studies have indicated that PPARs regulate the factors involved in fatty acid oxidation in the liver, and that PPAR-γ plays an important role in lipogenesis [14–16]. PPAR-γ is involved in the expression of lipogenic enzymes [17]. The herbal materials in HVC1, namely Coptidis Rhizoma (CR, rhizome of Coptischinensis), Scutellariae Radix (SR, radix of Scutellariabaicalensis), Rhei Rhizoma (RR, rhizome of Rheum officinale), and Pruni Cortex (PC, cortex of Prunusyedoensis Matsumare used as traditional medicines. The main bioactive compounds of CR are coptisine which improved obesity-related inflammatory response in syrian golden hamsters [18]. CR extract is reported to reduce oxidative stress and cholesterol levels [19] and the bioflavonoids from SR suppress the level of serum lipid [20] and RR possess hypolipidemic effects in hyperlipidemic rat model [21]. Among the components of HVC1, PC is reported to effective in cough, urticarial, dermatitis, and asthma [22, 23]. In addition, in our previous study, we found that prunetin, a component of Pruni Cortex, indicated anti-obesity effect via suppressing of adipogenesis [24].
In this study, we investigated HVC1’s potential to suppress high-cholesterol diet (HCD)-induced hyperlipidemia, and explored the possible molecular mechanism involved in the attenuation of lipid metabolism and hepatic inflammation.
Methods
Reagents
Monoclonal antibodies were purchased from Santa Cruz Biotechnology (California, USA). Oligonucleotide primers were purchased from Bioneer (Daejeon, Republic of Korea). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Preparation of HVC1
Pruni Cortex and Rhei Rhizome were purchased from Dongwoodang Co., Ltd. (Yeongcheon, Kyungpook, Republic of Korea). Coptidis Rhizoma and Scutellariae Radix were purchased from Dong Yang Herb Co., Ltd. (Seoul, Republic of Korea) [25]. The herbs were used at a ratio of 1:1:2:2 (CR 300 g: SR 300 g: PC 600 g: RR 600 g) respectively. The herbs were extracted using 30% (v/v) ethanol in water at 60 °C for 8 h. The extracts were filtered through a 10 μm cartridge paper, and the ethanol was removed by vacuum rotary evaporation (EYELA, Tokyo, Japan). The concentrates were freeze-dried, and the yield was calculated to be 13%. The powders were dissolved in distilled water for the experiments, and the residual powder was stored at −20 °C.
Experimental animals
LDL−/− mice (4 weeks old, male) were purchased from Daehan Biolink Co. Ltd. (Daejeon, Republic of Korea) and maintained under constant conditions (temperature, 22 ± 3 °C; humidity, 40–50%; light/dark cycle 12/12 h). Mice were adapted to the feeding conditions for 1 week and then given free access to food and tap water for 13 weeks. Mice were randomly separated into groups of 6 each (Table 1): ND (normal diet) group, HCD (high-cholesterol diet) group, and treatment groups fed HCD (D12336) and treated with simvastatin (10 mg/kg, p.o.) or HVC1 (10, 50, or 250 mg/kg, p.o.). With the exception of the ND group, all of the mice were fed a HCD. Body weight and dietary intake were recorded every week. At the last day of 13th week, the animals were fasted overnight. Blood samples were collected by cardiac puncture. The liver was excised, rinsed and directly stored at −80 °C until analyses. All procedures were conducted in accordance with the National Institute of Health guidelines and approved by the Ethical Committee for Animal Care and the Use of Laboratory Animals of Sangji University (reg.no. 2014–04).
Histological examination
To analysis of atherosclerosis, aorta roots were frozen in embedding media for Oil red O staining analysis. The aorta roots were sectioned at a thickness of 7 μm at −20 °C by using a CM3050 cryostat (Leica, Wetzlar, Germany). The slides were fixed and stained with Oil red O dye (sigma). After staining, the slides were washed three times with 1, 2-propranediol (85%) and then with deionized water. Images were acquired using an SZX10 microscope (Olympus, Tokyo, Japan). The fold change of Oil red O positive area was quantified using Adobe Photoshop 9.0.
Analysis of serum lipid profiles
Blood samples were collected and centrifuged at 1003 × g, for 15 min at room temperature to obtain serum samples. Unused samples were immediately frozen at −70 °C for later measurements. Serum concentrations of total cholesterol (TC), LDL cholesterol, HDL-cholesterol and triglycerides were determined by enzymatic methods with commercial kits (BioVision, Milpitas, California, USA).
Western blot analysis
Liver tissues were homogenized in PRO-PREP™ protein extraction solution (Intron Biotechnology, Seoul, Republic of Korea) and incubated for 20 min at 4 °C. Debris was removed by micro-centrifugation11000 xg, followed by quick freezing of the supernatants. The protein concentration was determined using the Bio-Rad protein assay reagent according to the manufacturer’s instructions (Bio-Rad, California, USA). Proteins were electro-blotted onto a polyvinylidene difluoride (PVDF) membrane following separation on a 10–12% SDS polyacrylamide gel. The membrane was incubated for 1 h with blocking solution (5% skim milk) at room temperature, followed by incubation overnight with primary antibodies including, PPAR-γ, SREPB-2, HMGCR, phosphor-AMPK, AMPK (dilution, 1:1000), β-actin (dilution, 1:2000) at 4 °C. Blots were washed three times with Tween 20/Tris-buffered saline (T/TBS) and incubated in a horseradish peroxidase-conjugated secondary antibody (dilution, 1:2500) for 2 h at room temperature. The blots were again washed three times with T/TBS, and then developed by enhanced chemiluminescence (GE Healthcare, Wisconsin, USA).
RNA separation and quantitative real-time PCR (qRT-PCR) analysis
The liver tissues were homogenized, and total RNA was isolated using a Trizol reagent (Invitrogen, Carlsbad, California, USA). cDNA was obtained using isolated total RNA (1 μg), d(T)16 primer, and AMV reverse transcriptase. Relative gene expression was quantified using real-time PCR (Real Time PCR System 7500, Applied Biosystems, California, USA) with SYBR green PCR master mix (Applied Biosystems). The gene Ct values of PPARs, HMGCR, SREBP-2, apoB, LPL, LXR and inflammatory cytokines were normalized using gene express 2.0 program (Applied Biosystems) to the Ct value of GAPDH. Oligonucleotide primers were purchased from Bioneer (Deajeon, Republic of Korea) (Table 2). The total reaction volume was 20 μl and consisted of 1 μl cDNA, 0.4 μl of primers, 10 μl SYBR green master mix and 8.2 μl DEPC treated water (Intron Biotechnology, Seoul, Republic of Korea).
Statistical analysis
Data are expressed as mean ± standard deviation (SD) of triplicate experiments. Statistically significance was determined using ANOVA and Dunnett’s post hoc test, and P-values of less than 0.05 were considered statistically significant.
Results
Effects of HVC1 on plaque formation and serum lipid profiles in HCD-fed LDLR−/− mice
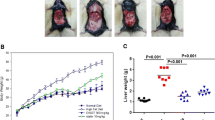
To investigate the effect of HVC1 on atherosclerosis, we examined Oil red O-stained lesions in the aortic roots. The plaque formation indicated as relative fold change of Oil red O positive area in HCD-fed LDLR−/− mice was almost 1.6 times higher than that in the ND group. In contrast to the HCD group, HVC1 administration significantly suppressed the plaque formation in dose-dependent manner in LDLR−/− mice fed HCD (Fig. 1a and b).

Effect of HVC1 on plaque formation in LDLR−/− mice fed a HCD. (a) Representative images of the aortic root and (b) quantitative analysis of Oil red O positive area. Oil red O staining was performed to show lipid in aortic root (black arrows, original magnification × 100). The fold change of Oil red O positive area was quantified using Adobe Photoshop 9.0. ND: Normal diet group; HCD: High-cholesterol diet group; Sim: Simvastatin (10 mg/kg) treated with HCD group; HVC1: HVC1 treated with HCD group. The values represent mean ± S.D. (significant as compared to HCD, ** p < 0.01, *** p < 0.001, significant as compared to ND, # p < 0.05)
After 13 weeks on HCD, the serum levels of TC, TG, and LDL-cholesterol in the HCD group were significantly higher than those in any other group (Fig. 2a-c). However, serum TC, TG and LDL-cholesterol levels in the HVC1-treated group (250 mg/kg, p.o.) significantly decreased compared to that in the HCD group. In addition, the HVC1 treated group (250 mg/kg) had increased levels of HDL-cholesterol when compared with the HCD group (Fig. 2d).

Effect of HVC1 on the serum lipid profile in LDLR−/− mice fed a HCD. Total-cholesterol (a), triglyceride (b), LDL-cholesterol (c), HDL-cholesterol (d). ND: Normal diet group; HCD: High-cholesterol diet group; Sim: Simvastatin (10 mg/kg) treated with HCD group; HVC1: HVC1 treated with HCD group. The values represent mean ± S.D. (significant as compared to HCD, * p < 0.05, *** p < 0.001, significant as compared to ND, ### p < 0.001)
Inhibitory effects of HVC1 on expression of PPARs in HCD-fed LDLR−/− mice
The effects of HVC1 on the mRNA expression of PPAR family members in liver tissue were examined by qRT-PCR analysis. As shown in Fig. 3, the mRNA expression of PPAR-α in the HCD group was not different from that in the ND group. However, in the HVC1 groups (10, 50, and 250 mg/kg, p.o.), the mRNA expression of PPAR-α significantly increased compared with the HCD group. In addition, administration of HVC1 (250 mg/kg, p.o.) increased the mRNA expression levels of PPAR-δ compared to that in the HCD group, while in the HCD group, PPAR-δ levels significantly decreased compared with the ND group. Conversely, the mRNA expression of PPAR-γ in the HVC1 group significantly decreased compared to that in the HCD group while being significantly higher in the HCD group than in the ND group. Since PPAR-γ plays an important role in adipogenesis and PPAR-γ activation results in reduction of free fatty acid (FFA) efflux and triacylglycerol synthesis [26], we examined PPAR-γ protein expression levels if the expression of PPAR-γ protein paralleled the transcription of its mRNA in LDLR−/− mice. As shown in Fig. 3d, PPAR-γ protein levels increased in the HCD group relative to the ND group. Compared to the HCD group, however, the simvastatin and HVC1-treated groups exhibited marked decrease in the protein levels of PPAR-γ.

Inhibitory effect of HVC1 on the HCD-induced PPARs expressions in LDLR−/− mice. LDLR−/− mice were randomized into the ND group, HCD group, and treatment groups fed HCD with simvastatin (10 mg/kg) or HVC1 (10, 50 or 250 mg/kg) for 13 weeks. mRNA levels of PPARs (a-c) were analyzed by real-time PCR analysis. Expressions of PPAR-γ protein was determined by western blot assay using specific anti-PPAR-γ antibody (d). β-actin was used as a loading control. Values represent mean ± S.D. of three independent experiments (significant as compared to HCD, * p < 0.05, ** p < 0.01, *** p < 0.001, significant as compared to ND, ### p < 0.001)
HVC1 regulates lipid metabolism and cholesterol synthesis in HCD-fed LDLR−/−mice
To investigate whether the lipid metabolism and biosynthesis of cholesterol in HVC1 treated mice was associated with molecular signaling by the genes involved in hyperlipidemia, we examined the expression levels of related genes that are key transcription factors in the liver. In the liver tissue, the mRNA expression levels of SREBP-2, HMGCR, LPL, and apo B significantly decreased in a dose dependent manner in the HVC1-treated groups compared to that in the HCD group. Conversely, the expression of LXR mRNA in the HVC1 groups increased compared to that in the HCD group (Fig. 4a-e). In addition, treatment with HVC1 markedly increased the expression levels of SREBP-2 and HMGCR in the HCD group compared to that in the ND group. The simvastatin and HVC1-treated groups, however, exhibited reduced expression levels of the proteins (Fig. 4f).

HVC1 regulation of cholesterol metabolism and lipid synthesis in LDLR−/− mice. Total RNA was subjected to real-time PCR as described in the Methods section. Cholesterol and lipid metabolism-related genes mRNA levels were analyzed by real-time PCR analysis. (a) SREBP-2, (b) HMGCR, (c) LPL, (d) apoB, and (e) LXR. Protein levels of (f) SREBP-2, (g) HMGCR, (H) p-AMPK, and AMPK in liver tissues were analyzed by western blot. Proteins were determined by western blot assay using specific antibody. β-actin was used as a loading control. Values represent mean ± S.D. of three independent experiments (significant as compared to HCD, * p < 0.05, ** p < 0.01, *** p < 0.001, significant as compared to ND, ### p < 0.001)
AMPK is a major regulator of energy metabolism, and its phosphorylation is involved in the regulation of adipocyte differentiation [27]; therefore, we investigated whether HVC1 regulated energy metabolism through the AMPK pathway. As shown in Fig. 4g, treatment with HVC1 inhibited HCD-induced dephosphorylation of AMPK in HCD-fed LDLR−/− mice.
Inhibitory effects of HVC1 on mRNA expression of inflammatory cytokines in HCD-fed LDLR−/− mice
Some studies have reported that a correlation exists between plasma total cholesterol and the development of hepatic inflammation [26, 28]. These reports led us to hypothesize that plasma TC as an important cause for the development of inflammation in HCD-fed LDLR−/− mice. Therefore, we investigated the expression of genes associated with inflammation in the liver of HCD-fed LDLR−/− mice. The mRNA expression levels of inflammatory cytokines in the HCD group were significantly up-regulated in comparison to those in the ND group. In contrast, HVC1 markedly reduced the mRNA expression of tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β in LDLR−/− mice (Fig. 5). The results suggest that HVC1 treatment may influence the HCD-induced expression of inflammatory genes in LDLR−/− mice.

Inhibitory effects of HVC1 on mRNA expression of inflammatory cytokines in HCD induced LDLR−/− mice. Inflammatory cytokine genes mRNA levels were analyzed by real-time PCR analysis. (a) TNF-α, (b) IL-1β, (c) IL-6.Total RNA was subjected to real-time PCR as described in the Methods section. Values represent mean ± S.D. of three independent experiments (significant as compared to HCD, * p < 0.05, ** p < 0.01, *** p < 0.001, significant as compared to ND, ### p < 0.001)
Discussion
Hyperlipidemia is related to increased levels of lipids, including cholesterol and triglyceride in the plasma. Hyperlipidemia increases the risk of developing cardiovascular disease [29]. In this study, we showed the inhibitory effect of HVC1 on hyperlipidemia-related factors in HCD-fed LDLR−/− mice. The effects of HVC1 included regulation of cholesterol synthesis, lipid accumulation, and levels of inflammatory cytokines in HCD-fed mice.
Increased blood lipid levels, especially LDL-cholesterol level, can promote atherosclerosis; therefore, decreasing lipid level is important in reducing atherosclerosis [30, 31]. In this study, HVC1 suppressed the serum levels of LDL-cholesterol, TG, and TC and increased HDL-cholesterol (Fig. 2). These results suggest that HVC1 is crucial for reducing the risk of hyperlipidemia.
PPARs are members of the nuclear hormone receptor superfamily, and they regulate various physiological functions, such as glucose and lipid homeostasis, inflammatory responses, cell differentiation [5]. Lee et al. have reported that PPAR-α and-γ agonists (WY14643 and rosiglitazone) decreased hyperlipidemia by increasing the protein expression of malonyl-CoA decarboxylase (MCD) [32]. PPAR-δ agonists are also reported to have a role in lipid metabolism and they improve metabolic syndrome. They have been noted to enhance HDL cholesterol and decrease LDL cholesterol in insulin-resistant obese monkeys [33]. PPAR-γ plays a role in hyperlipidemia, triglyceride clearance and hepatic steatosis [27, 34]. Some studies reported that HCD administration lead to hepatic damage such as hepatic steatosis in mice [35, 36] and PPAR-γ act as a key modulator of high-fat diet-induced liver steatosis [37, 38]. In this study, we found that HVC1 enhanced the mRNA expression levels of PPAR-α and δ, while HVC1 significantly decreased PPAR-γ mRNA and protein expression levels in liver which is induced to hepatic steatosis. These results suggested that modulation of PPARs expression might be one of the mechanisms by which HVC1attenuates lipid accumulation in hyperlipidemia and hepatic steatosis affected by HCD.
The liver plays a central role in lipid metabolism. Some orphan nuclear hormone receptors such as LXR, retinoid X receptor (RXR), farnesoid X receptor (FXR), and PPAR are related to genes involved in cholesterol metabolism [39, 40]. LXR plays a key important role in cholesterol sensor. LDLR−/− mice have functionally disordered bile acid production, which leads to cholesterol ester accumulation [41]. In addition, SREBP is a transcription factor that regulates the biosynthesis of cholesterol and fatty acids [42]. The precursor of SREBP is synthesized in the endoplasmic reticulum (ER) membrane-bound protein. It is activated, cleaved, and then translocated to the nucleus. SREBP-2 promotes the expression of target genes involved in cholesterol biosynthesis such as 3-hydroxy-3-methylglutaryl-CoA synthase (HMGCS), HMGCR and LDLR [43, 44]. HMGCR is a transmembrane protein that is associated with the biosynthesis of cholesterol [45, 46]. In this study, HVC1 decreased the mRNA expression levels of SREBP-2, HMGCR, LPL, and apoB and increased that of LXR in the liver tissue of HCD-fed LDLR−/− mice. In addition, HVC1 markedly increased the protein expression level of SREBP-2 and HMGCR. These data suggested that HVC1 regulated lipid synthesis-related markers through the modulation of adipogenic gene related to cholesterol metabolism.
AMPK is a complex of α/β/γ subunits, which regulate lipid and carbohydrate metabolism, immune response, cell growth, and protein synthesis [47, 48]. AMPK plays an important role in the regulation of fatty acid oxidation by the phosphorylation and inactivation of acetyl-CoA carboxylase (ACC) [49]. It also plays a central role in lipid metabolism in the liver. Hepatic ACC has been found to be regulated in the liver [50]. In addition, AMPK is a key enzyme in cholesterol synthesis via regulation HMGCR [51]. Therefore, AMPK regulates fatty acid oxidation and cholesterol synthesis. The activation of AMPK, results in suppression of lipogenesis in the liver, which inhibits lipid accumulation [52, 53] and AMPK phosphorylation is inhibited in mice fed a HCD [27]. In this study, HVC1 significantly increased AMPK phosphorylation resulting in regulation of lipid metabolism-related genes in HCD-fed LDLR−/−mice (Fig. 4). These findings suggested that hypolipidemic effects of HVC1 could dependent on AMPK pathway in HCD-fed LDLR−/−mice.
Hyperlipidemia not only involves elevation in serum lipids, but it is also an inflammatory disease, as excessive lipid accumulation has been known to trigger local inflammatory reactions [54, 55]. The inflammatory processes mostly coincide with increased local fat accumulation as observed in nonalcoholic steatohepatitis [14]. In addition, inflammatory processes occur during the cardiovascular disease process [33, 56, 57]. Blockade of inflammatory cytokines has been shown to reduce the incidence of cardiovascular disease [29]. In this study, our results showed that HVC1 significantly reduced the mRNA expression levels of inflammatory cytokines (TNF-α, IL-1β, and IL-6) in HCD-fed LDLR−/− mice. Many cytokines participated in the development of atherosclerotic leading to plaque formation [58]. Expression of IL-1-family members and their receptors has been demonstrated in atherosclerotic plaques. Furthermore, inhibition of TNF-α reduces atherosclerosis in apolipoprotein E knockout mice [59]. In these regards, we could suggest that inhibitory effects of HVC1 on mRNA expression levels of inflammatory cytokines are related to the development of atherosclerotic plaque.
Conclusions
We demonstrated that HVC1 has an inhibitory effect of hyperlipidemia involving inflammation in HCD-fed LDLR−/− mice. Our findings indicated that HVC1 treatment could suppress the development of hyperlipidemia via regulation of cholesterol metabolism and inflammatory processes as observed in the HCD-fed LDLR−/− mice. Therefore, HVC1 may be used for the prevention or treatment of hyperlipidemia.
Abbreviations
- LDLR−/− :
-
Low-density lipoprotein receptor-deficient
- TG:
-
Triglyceride
- LDL:
-
Low-density lipoprotein
- HDL:
-
High-density lipoprotein
- PPARs:
-
Peroxisome proliferator-activated receptors
- SREBP-2:
-
Sterol regulatory element-binding proteins
- HMG-CoA reductase:
-
3-hydroxy-3-methylglutaryl
- LPL:
-
Lipoprotein lipase
- apo B:
-
Apolipoprotein B
- LXR:
-
Liver X receptor
- AMPK:
-
5′ adenosine monophosphate-activated protein kinase
- HCD:
-
High-cholesterol diet
- FFA:
-
Free fatty acid
- TNF:
-
Tumor necrosis factor
- IL:
-
Interleukin
- MCD:
-
Malonyl-CoA decarboxylase
- ACC:
-
Acetyl-CoA carboxylase
References
Le NA. Hyperlipidemia and cardiovascular disease. Curr Opin Lipidol. 2006;17(6):702–4.
Gotto Jr AM. Jeremiah Metzger lecture: cholesterol, inflammation and atherosclerotic cardiovascular disease: is it all LDL? Trans Am Clin Climatol Assoc. 2011;122:256–89.
Brown MS, Goldstein JL. A receptor-mediated pathway for cholesterol homeostasis. Science. 1986;232(4746):34–47.
Ishibashi S, Brown MS, Goldstein JL, Gerard RD, Hammer RE, Herz J. Hypercholesterolemia in low density lipoprotein receptor knockout mice and its reversal by adenovirus-mediated gene delivery. J Clin Invest. 1993;92(2):883–93.
Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J Clin Invest. 2006;116(3):598–606.
Takano H, Hasegawa H, Zou Y, Komuro I. Pleiotropic actions of PPAR gamma activators thiazolidinediones in cardiovascular diseases. Curr Pharm Des. 2004;10(22):2779–86.
Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20(5):649–88.
Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cellular and molecular life sciences : CMLS. 2004;61(4):393–416.
Francis GA, Annicotte JS, Auwerx J. PPAR-alpha effects on the heart and other vascular tissues. Am J Physiol Heart Circ Physiol. 2003;285(1):H1–9.
Ory DS. Nuclear receptor signaling in the control of cholesterol homeostasis: have the orphans found a home? Circ Res. 2004;95(7):660–70.
Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M, Grimaldi PA. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2003;17(15):2299–301.
Oliver Jr WR, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci U S A. 2001;98(9):5306–11.
Naya N, Fukao K, Nakamura A, Hamada T, Sugimoto M, Kojima M, Yoshimura N, Uwabe K, Imagawa K, Nomura K, et al. A selective peroxisome proliferator-activated receptor delta agonist PYPEP suppresses atherosclerosis in association with improvement of the serum lipoprotein profiles in human apolipoprotein B100 and cholesteryl ester transfer protein double transgenic mice. Metab Clin Exp. 2016;65(1):16–25.
Stienstra R, Mandard S, Patsouris D, Maass C, Kersten S, Muller M. Peroxisome proliferator-activated receptor alpha protects against obesity-induced hepatic inflammation. Endocrinology. 2007;148(6):2753–63.
Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, Suzuki Y, Saito H, Kohgo Y, Okumura T. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336(1):215–22.
Furnsinn C, Willson TM, Brunmair B. Peroxisome proliferator-activated receptor-delta, a regulator of oxidative capacity, fuel switching and cholesterol transport. Diabetologia. 2007;50(1):8–17.
Walczak R, Tontonoz P. PPARadigms and PPARadoxes: expanding roles for PPARgamma in the control of lipid metabolism. J Lipid Res. 2002;43(2):177–86.
Zou ZY, Hu YR, Ma H, Wang YZ, He K, Xia S, Wu H, Xue DF, Li XG, Ye XL. Coptisine attenuates obesity-related inflammation through LPS/TLR-4-mediated signaling pathway in Syrian golden hamsters. Fitoterapia. 2015;105:139–46.
Yokozawa T, Ishida A, Cho EJ, Nakagawa T. The effects of Coptidis Rhizoma extract on a hypercholesterolemic animal model. Phytomedicine : international journal of phytotherapy and phytopharmacology. 2003;10(1):17–22.
Regulska-Ilow B, Biernat J, Grajeta H, Ilow R, Drzewicka M. Influence of bioflavonoids from the radix extract of Scutellaria Baicalensis on the level of serum lipids, and the development of laboratory rats fed with fresh and oxidized fats. Die Nahrung. 2004;48(2):123–8.
Kim YS, Jung EA, Shin JE, Chang JC, Yang HK, Kim NJ, Cho KH, Bae HS, Moon SK, Kim DH. Daio-Orengedokuto inhibits HMG-CoA reductase and pancreatic lipase. Biol Pharm Bull. 2002;25(11):1442–5.
Ahn DK: Illustrated book of Korean medicinal herbs: Kyohak; 2006.
Kim JG. Illustrated Natural Drugs Encyclopedia (color edition) Vol.1. Seoul: Namsandang; 1992.
Ahn TG, Yang G, Lee HM, Kim MD, Choi HY, Park KS, Lee SD, Kook YB, An HJ. Molecular mechanisms underlying the anti-obesity potential of prunetin, an O-methylated isoflavone. Biochem Pharmacol. 2013;85(10):1525–33.
Lee K, Kim B, Hur H, Chinannai KS, Ham I, Choi HY. Antihypertensive effect of the GaMiSamHwangSaSimTang in spontaneous hypertensive rats. Evidence-based complementary and alternative medicine : eCAM. 2015;2015:802368.
Wouters K, van Gorp PJ, Bieghs V, Gijbels MJ, Duimel H, Lutjohann D, Kerksiek A, van Kruchten R, Maeda N, Staels B, et al. Dietary cholesterol, rather than liver steatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholic steatohepatitis. Hepatology. 2008;48(2):474–86.
Ejaz A, Wu D, Kwan P, Meydani M. Curcumin inhibits adipogenesis in 3T3-L1 adipocytes and angiogenesis and obesity in C57/BL mice. J Nutr. 2009;139(5):919–25.
Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, Garcia-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4(3):185–98.
Jacobsson LT, Turesson C, Gulfe A, Kapetanovic MC, Petersson IF, Saxne T, Geborek P. Treatment with tumor necrosis factor blockers is associated with a lower incidence of first cardiovascular events in patients with rheumatoid arthritis. J Rheumatol. 2005;32(7):1213–8.
Arsenault BJ, Rana JS, Stroes ES, Despres JP, Shah PK, Kastelein JJ, Wareham NJ, Boekholdt SM, Khaw KT. Beyond low-density lipoprotein cholesterol: respective contributions of non-high-density lipoprotein cholesterol levels, triglycerides, and the total cholesterol/high-density lipoprotein cholesterol ratio to coronary heart disease risk in apparently healthy men and women. J Am Coll Cardiol. 2009;55(1):35–41.
Durrington P. Dyslipidaemia Lancet. 2003;362(9385):717–31.
Luchtefeld M, Grothusen C, Gagalick A, Jagavelu K, Schuett H, Tietge UJ, Pabst O, Grote K, Drexler H, Forster R, et al. Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation. 2010;122(16):1621–8.
Tietge UJ. Hyperlipidemia and cardiovascular disease: inflammation, dyslipidemia, and atherosclerosis. Curr Opin Lipidol. 2014;25(1):94–5.
Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278(36):34268–76.
Ganz M, Csak T, Szabo G. High fat diet feeding results in gender specific steatohepatitis and inflammasome activation. World J Gastroenterol. 2014;20(26):8525–34.
Lee HJ, Yeon JE, Ko EJ, Yoon EL, Suh SJ, Kang K, Kim HR, Kang SH, Yoo YJ, Je J, et al. Peroxisome proliferator-activated receptor-delta agonist ameliorated inflammasome activation in nonalcoholic fatty liver disease. World J Gastroenterol. 2015;21(45):12787–99.
Memon RA, Tecott LH, Nonogaki K, Beigneux A, Moser AH, Grunfeld C, Feingold KR. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology. 2000;141(11):4021–31.
Bedoucha M, Atzpodien E, Boelsterli UA. Diabetic KKAy mice exhibit increased hepatic PPARgamma1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J Hepatol. 2001;35(1):17–23.
Repa JJ, Mangelsdorf DJ. Nuclear receptor regulation of cholesterol and bile acid metabolism. Curr Opin Biotechnol. 1999;10(6):557–63.
Russell DW. Nuclear orphan receptors control cholesterol catabolism. Cell. 1999;97(5):539–42.
Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXR alpha. Cell. 1998;93(5):693–704.
Matsuda M, Korn BS, Hammer RE, Moon YA, Komuro R, Horton JD, Goldstein JL, Brown MS, Shimomura I. SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 2001;15(10):1206–16.
Goldstein JL, DeBose-Boyd RA, Brown MS. Protein sensors for membrane sterols. Cell. 2006;124(1):35–46.
Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–40.
Bloomfield DK. Dynamics of cholesterol metabolism. I Factors regulating total sterol biosynthesis and accumulation in the rat Proceedings of the National Academy of Sciences of the United States of America. 1963;50:117–24.
Bonne AC, den Bieman MG, Gillissen GF, van Lith HA, van Zutphen LF. Chromosomal localization of genes involved in biosynthesis, metabolism or transport of cholesterol in the rat. Cytogenetic and genome research. 2002;97(3–4):183–6.
Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8(10):774–85.
Steinberg GR, Macaulay SL, Febbraio MA, Kemp BE. AMP-activated protein kinase--the fat controller of the energy railroad. Can J Physiol Pharmacol. 2006;84(7):655–65.
Ouchi N, Shibata R, Walsh K. AMP-activated protein kinase signaling stimulates VEGF expression and angiogenesis in skeletal muscle. Circ Res. 2005;96(8):838–46.
Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci U S A. 2002;99(25):15983–7.
Hayashi T, Hirshman MF, Kurth EJ, Winder WW, Goodyear LJ. Evidence for 5′ AMP-activated protein kinase mediation of the effect of muscle contraction on glucose transport. Diabetes. 1998;47(8):1369–73.
Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1(1):15–25.
Wu CH, Yang MY, Chan KC, Chung PJ, Ou TT, Wang CJ. Improvement in high-fat diet-induced obesity and body fat accumulation by a Nelumbo nucifera leaf flavonoid-rich extract in mice. J Agric Food Chem. 2010;58(11):7075–81.
van der Heijden RA, Bijzet J, Meijers WC, Yakala GK, Kleemann R, Nguyen TQ, de Boer RA, Schalkwijk CG, Hazenberg BP, Tietge UJ, et al. Obesity-induced chronic inflammation in high fat diet challenged C57BL/6J mice is associated with acceleration of age-dependent renal amyloidosis. Sci Rep. 2015;5:16474.
Otogawa K, Kinoshita K, Fujii H, Sakabe M, Shiga R, Nakatani K, Ikeda K, Nakajima Y, Ikura Y, Ueda M, et al. Erythrophagocytosis by liver macrophages (Kupffer cells) promotes oxidative stress, inflammation, and fibrosis in a rabbit model of steatohepatitis: implications for the pathogenesis of human nonalcoholic steatohepatitis. Am J Pathol. 2007;170(3):967–80.
Navab KD, Elboudwarej O, Gharif M, Yu J, Hama SY, Safarpour S, Hough GP, Vakili L, Reddy ST, Navab M, et al. Chronic inflammatory disorders and accelerated atherosclerosis: chronic kidney disease. Curr Pharm Des. 2011;17(1):17–20.
Fitzgerald ML, Mujawar Z, Tamehiro N. ABC transporters, atherosclerosis and inflammation. Atherosclerosis. 2010;211(2):361–70.
Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31(5):969–79.
Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S. Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. 2004;24(11):2137–42.
Acknowledgments
This study was supported by a Grant from the Korea Health care Technology R&D Project, Ministry of Health & Welfare, Republic of Korea (B110081).
Availability of data and materials
Data are all contained within the article.
Authors’ contributions
HJA conceived and designed the study. SYC and KSC carried out the experiments, analyzed the research data, and wrote the manuscript. KJL, HYC and IHH participated in the study design and analyzed the research data. DHJ and YYC contributed essential reagents. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cheon, SY., Chung, KS., Lee, KJ. et al. HVC1 ameliorates hyperlipidemia and inflammation in LDLR−/− mice. BMC Complement Altern Med 17, 222 (2017). https://doi.org/10.1186/s12906-017-1734-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12906-017-1734-z