
Abstract
Background
The aim of this study was to analyse the differences in the phenotypes of missing teeth between a pair of brothers with hypohidrotic ectodermal dysplasia (HED) and to investigate the underlying mechanism by comparing the mutated gene loci between the brothers with whole-exome sequencing.
Methods
The clinical data of the patients and their mother were collected, and genomic DNA was extracted from peripheral blood samples. By Whole-exome sequencing filtered for a minor allele frequency (MAF) ≤0.05 non-synonymous single-nucleotide variations and insertions/deletions variations in genes previously associated with tooth agenesis, and variations considered as potentially pathogenic were assessed by SIFT, Polyphen-2, CADD and ACMG. Sanger sequencing was performed to detect gene variations. The secondary and tertiary structures of the mutated proteins were predicted by PsiPred 4.0 and AlphaFold 2.
Results
Both brothers were clinically diagnosed with HED, but the younger brother had more teeth than the elder brother. An EDA variation (c.878 T > G) was identified in both brothers. Additionally, compound heterozygous variations of WNT10A (c.511C > T and c.637G > A) were identified in the elder brother. Digenic variations in EDA (c.878 T > G) and WNT10A (c.511C > T and c.637G > A) in the same patient have not been reported previously. The secondary structure of the variant WNT10A protein showed changes in the number and position of α-helices and β-folds compared to the wild-type protein. The tertiary structure of the WNT10A variant and molecular simulation docking showed that the site and direction where WNT10A binds to FZD5 was changed.
Conclusions
Compound heterozygous WNT10A missense variations may exacerbate the number of missing teeth in HED caused by EDA variation.
Similar content being viewed by others


Introduction
Tooth agenesis (TA) is divided into nonsyndromic and syndromic types according to the presence or absence of multiple clinical syndromes [1, 2]. Nonsyndromic tooth agenesis (NSTA) is by far the most common form of tooth agenesis [2]. NSTA presents as an isolated trait that only affects dentition. Epidemiological studies indicate that the prevalence of NSTA ranges from 2.6 to 11.3% in various nations and races [3,4,5]. Numerous genes account for the aetiology of NSTA, MSX1, PAX9, AXIN2, WNT10A, WNT10B, LRP6, EDA are most frequently mentioned in relationship to TA [6,7,8,9,10,11,12]. WNT10A, which is located on chromosome 2q35, has been reported as a major pathogenic gene suggested to account for as much as 50% of NSTA cases [13, 14].
TA can also be part of a complex disorder in a syndromic form, which is known as syndromic tooth agenesis, such as ectodermal dysplasia (EDs). EDs are classified into 11 clinical subgroups, and hypohidrotic ectodermal dysplasia (HED) is one of the most common type of EDs [2, 15, 16], presenting with hypodontia/oligodontia, hypohidrosis/anhidrosis, and hypotrichosis [16,17,18]. Additional dysmorphic features may be associated with HED, including a prominent forehead, rings under the eyes, saddle nose, prominent protruding lips, periocular pigmentation, alterations in the meibomian glands, and the sporadic absence of nipples [18,19,20]. Four genes are known to cause about 90% of hypohidrotic/anhidrotic ED cases to date: EDA, EDAR, EDARADD, and WNT10A [18]. The majority of HED cases are associated with variations or deletions in EDA gene (Xq12-q13.1) inherited on the X-chromosome. The WNT10A gene was determined to be responsible for various autosomal recessive forms of EDs, including onycho-odonto-dermal dysplasia (OODD) and Schöpf-Schulz-Passarge syndrome [21, 22]. Moreover, recent studies revealed that WNT10A gene variations could also cause HED [18, 22, 23]. Compared to HED cases caused by EDA variations, the clinical phenotype of HED caused by WNT10A variations is milder, exhibiting abnormal hair and sweat glands but no facial deformities [18]. And the remaining 10% of hypohidrotic/anhidrotic ED cases caused by rarer genetic variations, such as NFKBIA [24,25,26], and NEMO/IKBKG [27,28,29]. However, the molecular mechanisms and signalling pathways underlying HED with WNT10A variations have not been fully elucidated and the correlation between the number and location of missing teeth in HED and the pathogenic genes has not yet been fully elucidated.
In our clinical study, a pair of brothers were diagnosed with HED, and both had the characteristic features of hypodontia, hypotrichosis, hypohidrosis, and facial dysmorphism. Interestingly, the dental hypoplasia of the elder brother was much more severe than that of the younger brother. The elder brother presented with the mandibular edentulous jaw and only two maxillary central incisors, while the younger brother had some anterior teeth. To study the genetic pathogenesis, whole-exome sequencing (WES) was performed. The results revealed that the elder brother had both EDA and WNT10A variations, while his younger brother only had the EDA variation. To explore whether the mutated WNT10A protein had impaired function, we predicted its three-dimensional structure and analysed the functional changes in the mutated protein.
Materials and methods
Clinical data collection
Two brothers (11-year-old and 8-year-old) were referred to the Center of Stomatology, Xiangya Hospital, Central South University (China), complaining of congenital missing teeth. With informed consent from the mother and the brothers, we collected the patients’ medical history, took photos, and drew peripheral venous blood samples.
Whole-exome sequencing (WES) and analysis of sequencing results
DNA extraction and WES of qualified DNA samples were performed by Genergy Bio-Technology (Shanghai) Co., LTD.. According to the WES results, firstly, we removed any variation based on a minor allele frequency (MAF) > 0.05 in any of the three database: 1000 Genomes (1000G; https://www.internationalgenome.org/), Exome Aggregation Consortium (ExAC; http://exac.broadinstitute.org/) or Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/), all of which contain normal healthy individuals from the East Asian population who were similar to the patients [30, 31]. Secondly, we removed synonymous single-nucleotide, and filtered all nonsynonymous single-nucleotide variants and insertions/deletions located in the exon regions (Supplementary Table 1). Then, we filtered variations in genes from the list of 55 genes associated with TA reported in previous studies (Supplementary Table 2). Additionally, based on the scoring criteria of the following tools:(1) SIFT score ≤ 0.05, (2) Polyphen-2 score ≥ 0.909, (3) CADD score > 20, (4) meeting ACMG criteria, we identified potential pathogenicity of reserved variations associated with TA.
Sanger sequencing
To verify the WES results, the related EDA and WNT10A fragments were sequenced using Sanger sequencing. Genomic DNA from the brothers was isolated according to the procedure using a the TIANamp Blood DNA Midi Kit (Tiangen, Beijing, China) according to the manufacturer’s procedure. The primers used were specifically designed to detect the variations (Table 1). The coding sequences of the EDA and WNT10A genes were amplified using PCR with Taq PCR Master Mix (BioTek, Beijing, China). The PCR products were sequenced by Tsingke Biotechnology Co., Ltd. (Beijing). The results were compared with the reference sequences for each gene (EDA, NM_001399; WNT10A, NM_025216) (UCSC, http://genome.ucsc.edu/) to verify the results of WES.
Structure prediction
The National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) was used to query the original amino acid sequences encoded by the disease-related genes. The secondary structures of the wild-type and mutated variants of EDA and WNT10A were predicted using PsiPred 4.0 (http://bioinf. cs.ucl.ac.uk/psipred). Homology modelling analysis and tertiary structure prediction were performed with AlphaFold 2 (https://www.alphafold.ebi.ac.uk/). HDOCK, a web server of molecular docking (http://hdock.phys.hust.edu.cn/), was used to predict of protein-protein interactions. The structures of the wild-type and mutant proteins were generated and compared by PyMOL software (PyMOL Molecular Graphics System; DeLano Scientific).
Results
The elder brother has a more severe phenotype of tooth agenesis than the younger brother
Physical examination showed that both brothers presented sparse hair, missing teeth and sweat gland dysplasia (Fig. 1a-d). They both exhibited the typical facial appearance of X-linked HED: saddle nose, prominent thick lips, a pointed chin, and rings under the eyes. Oral examination and cone beam computed tomography (CBCT) examination showed that all primary teeth and most permanent teeth of the elder brother (II-1) were absence congenitally. Only 2 peg-shaped teeth (#11,21) were remained (Fig. 1a-b). Due to the total loss of mandibular dentition, he was unable to chew or construct occlusion. The younger brother (II-2) still had 6 primary teeth (#51,53,61,63,73,83) and 3 permanent teeth germs (#11,21,43) (Fig. 1c-d). Considering the systematic and oral manifestations, both the two brothers were diagnosed with HED.

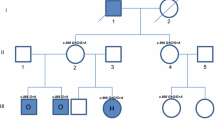
Dental characteristics and facial features of the pair of brothers with HED. a-b Oral conditions, panoramic radiographs of the proband (II-1). c-d Oral conditions, panoramic radiographs of the little brother (II-2). e. Pedigree structure of the family with HED, and black squares represent HED patients. f. DNA sequencing chromatogram showing a heterozygous EDA variant of c.878 T > G (p.L293R) in the pair of brothers (II-1, II-2). g-h Two heterozygous WNT10A variants of c.511C > T (p.R171C) and c.637G > A (p.G213S) in the proband (II-1)
A missense variation of EDA c.878 T > G (p.L293R) was identified in both brothers, but compound heterozygous WNT10A variations (c.511C > T (p.R171C) and c.637G > A (p.G213S)) were found only found in the elder brother
WES screening revealed 3 missense variations in the elder brother, including c.878 T > G (p.L293R) in exon 7 of the EDA gene and c.511C > T (p.R171C) and c.637G > A (p.G213S) in exon 3 of the WNT10A gene. The three variations were predicted to be damaging by in silico tools (Table 2). The variation sites have been deposited in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/submitters/509028/). The WNT10A variations (c.511C > T and c.637G > A) were compound heterozygous variations identified in the pathogenesis of TA. The younger brother had only the missense variation of c.878 T > G (p.L293R) of EDA. Their mother is clinically unaffected and has none of the phenotypes involving hair, sweat glands, or teeth. WES screening revealed that she carries the EDA heterozygous variation (c.878 T > G) and one of the WNT10A heterozygous variations (c.511C > T). These results were confirmed them by Sanger sequencing (Fig. 1e-h).
The mutant protein structures WNT10A and EDA were predicted
The secondary and tertiary protein structures were predicted to analyse the effects of the variants on the protein functions. The secondary structures of the variant EDA (p.L293R) and WNT10A (p.R171C, p.G213S) proteins were predicted and demonstrated that these three missense variants could lead to changes in multiple α-helices and β-folds in the secondary structure of EDA and WNT10A proteins (Fig. 2a-e). Tertiary structure analysis of the EDA (p.L293R) protein showed that the length of the hydrogen bonds between amino acid 293 and amino acid 360 was changed, which results in an extra α-helix of the protein structure (Fig. 3d). In the variant WNT10A (p.R171C, p.G213S) protein, the leucine was replaced by the serine, and the length of the hydrogen bonds formed between the mutated amino acids and the surrounding amino residues was changed (Fig. 3a-b).

Secondary structure analysis of mutated proteins. a The predicted secondary structure of the wild-type EDA protein. b The predicted secondary structure of mutated EDA protein (p.L293R). c. The predicted secondary structure of the wild-type WNT10A protein. d. The predicted secondary structure of mutated WNT10A proteins (p.R171C). e. The predicted secondary structure of mutated WNT10A proteins (p.G213S). Sites of variants are indicated by green squares. The structural changes in these mutated proteins compared to the wild-type proteins (EDA and WNT10A) are indicated by orange squares. α-Helices are represented as pink squares, while coils are represented as grey squares

Prediction of tertiary structure of proteins. a-b Tertiary structure prediction and hydrogen bond analysis of WNT10A wild-type and WNT10A p.R171C and p.G213S. d Tertiary structure prediction and hydrogen bond analysis of EDA wild-type and EDA p.L293R. c, e (1)–(3). Simulation of molecular docking between WNT10A wild-type and FZD5, and three binding sites between them. c, e (1′)-(3′). Simulation of molecular docking between WNT10A (p.R171C and p.G213S) and FZD5, and three binding sites between them
FZD5 has been reported to interact with numerous WNT proteins including WNT10A, and to activate signalling cascades in various cellular and developmental processes [32,33,34]. To explore the possible mechanism of the altered WNT signal, we predicted the ligand-protein molecular docking of the compound heterozygous mutant (p.R171C and p.G213S) protein with FZD5. Compared with ligand-protein molecular docking of the wild-type WNT10A with the FZD5 α-helix, the WNT10A variant was found to mainly bind to the FZD5 loop domain (Fig. 3c). In addition, we revealed that both WNT10A wild-type and WNT10A variant had three binding sites with FZD5 (Fig. 3c, e). The binding sites of the wild-type WNT10A were located at the C-terminal thumb domain of FZD5, while the variant WNT10A not only binds to the C-terminal thumb domain of FZD5, but also to the N-terminal index finger domain of FZD5. The variation changed the position and direction of the binding site, resulting in a changed dimer structure formed by compound heterozygous mutant WNT10A and FZD5. It suggests compound heterozygous WNT10A mutants may have a negative effect on WNT signalling. In addition, we also predicted the binding of WNT10A (p.R171C) and WNT10A (p.G213S) to FZD5, respectively and we found that the dimers WNT10A (p.R171C) -FZD5 and WNT10A (p.G213S)-FZD5 were almost identical in structure to the wild-type WNT10A-FZD5 dimer (Fig. 3c, Supplementary Fig. 1a-b), although their binding sites were not exactly the same (Supplementary Fig. 1c-d).
Discussion
HED displays different modes of inheritance according to the gene that is involved, with X-linked EDA-related HED being the most frequent form of the disease. EDA is located in the Xq12-q13 region, which has multiple subtypes and mainly mediates epidermal-mesenchymal and cell–cell signalling transduction [35,36,37]. Classical EDA signaling components include EDA, EDAR and EDARDD, and the downstream signalling pathway is the NF-κB signalling pathway [38, 39]. The EDA gene has 4 important functional areas [40,41,42]: TM, Furin cleavage, Collagen-like domain (collagen) and TNF homology domain, the latter of which is the most commonly mutated structure [43]. The c.878 T > G variation found in our study [44], is located in the TNF homology domain of the EDA gene. The TNF homology domain binds to the receptor (EDAR) to form an autotrimer. We speculate that the c.878 T > G variation in the TNF homology domain prevents EDA from binding to EDAR and therefore affects EDA signal transduction, resulting in increased expression of IκBα that the most important member in inhibiting the activation of NF-κB [45, 46] by inhibiting the phosphorylation of NF-κB, nuclear transcription and binding to DNA, which is downstream of the NF-κB signalling pathway. The inactivation of the NF-κB signalling pathway leads to the developmental defects in ectoderm-derived tissues and organs [47, 48]. Our finding was similar to that of a previous study, in which a patient with the c.878 T > G variation had only two peg-shaped central incisors remaining in the upper jaw [44].
Large EDA variations identified in HED have been reported, and the phenotypes of these variations vary. In this study, we summarized the HED phenotypes caused by EDA variations in the past 5 years (Table 3). We found statistically that 43.14% (22/51) of EDA variations was located in the TNF homology subdomain (Fig. 4a), which is consistent with previous reports [43]. The average number of missing teeth caused by variations in different EDA domains was greater than 24, with no significant differences (Fig. 4c), which is consistent with the phenotype of the patients in this study. Different tooth positions were affected; the maxillary central incisor was the least affected, with a retention rate of 0.538 (14/26, #11 and #21), followed by left maxillary first molars with a retention rate of 0.385 (10/26, #26) (Fig. 4b). As shown in Table 3, patients with the c.457C > T variation all retained the upper and lower first molars, and patients with the c.164 T > C variation retained the maxillary central incisors. In addition, we found that patients with the same EDA variation had differences in missing tooth phenotypes, which may be caused by epigenetics or other variations that have not been detected.

Summary of molecular genetic data of EDA variations caused HED. a The distribution of various variation domains of EDA in HED patients. b The frequency of tooth retention caused by EDA variations at each dental position. c The average number of missing teeth caused by variations in each functional domain of EDA. There was no significant difference between groups (P > 0.5) by Kruskal-Wallis H test
WNT10A is considered to be a common pathogenic gene in NSTA, and is located at chromosome 2q35 [70]. By acting as a short-range ligand, WNT10A locally activates the receptor-mediated WNT signalling pathway [33]. WNT10A was found to be expressed in the dental epithelium and the enamel knot, as well as in the mesenchymal preodontoblast layer during tooth development and is required for dentinogenesis and tooth morphogenesis [71, 72]. WNT10A comprises an N-terminal index finger domain with α-helices (NTD) from residue 1 to residue 250, and a C-terminal cysteine-rich region (CTD) with a thumb from residue 261 to residue 338. WNT10A c.511C > T (p.R171C) and c.637G > A (p.G213S), located in exon 3, are the two frequent variations in Asian NSTA population [73, 74]. The compound heterozygous WNT10A variations (c.511C > T and c.637G > A) had been reported in a male who diagnosed with NSTA [75]. He was two teeth absent clinically, without any symptoms of ectodermal dysplasia [75]. The two variations are both located in the N-terminal index finger domain, near 4 disulfide bonds [34], and the variations of amino residues may destroy disulfide bond binding, affect the stability of protein structure, and eventually lead to destabilizing of the N-terminal index finger domain [34, 76]. Neither variation has been reported in cases of HED, although the of c.637G > A variation has been reported to cause minor signs of ED [77]. Currently, it is believed that the tooth loss phenotype caused by compound heterozygous WNT10A variations is far more serious than that caused by a single heterozygous WNT10A variation [78].
In this study, we collected cases of TA with concurrent EDA and WNT10A digenic variations (Table 4). In a previous study on TA by WNT10A and EDA digenic variations, patients with simple heterozygous WNT10A and EDA digenic variations had no more severe phenotypes than those previously reported with EDA single variations [78]. In this study, which compared a pair of biological brothers, may have excluded the influence of other confounding factors. We found that the presence of two variations (c.511C > T and c.637G > A) in WNT10A led to a more severe absence of teeth in HED patients.
The EDA/EDAR/NF-κB signalling pathway is now recognized as a classical signalling pathway associated with the incidence of HED [16, 18]. Studies have shown the EDA/EDAR/NF-κB signalling pathway is closely related to the Wnt/β-catenin signalling pathway, and they regulate each other. During hair follicle development, Wnt/β-catenin signaling is required for NF-κB activation, while EDAR/NF-κB subsequently enhances and maintains Wnt/β-catenin activity [79]. Meanwhile, during tooth morphogenesis, Wnt10A and EDAR are both expressed in the dental epithelium at initiation and bud stages and in the enamel knot in the cap stage [76]. Wnt signals regulate ectodysplasin expression in the oral ectoderm, and EDAR expression in the epithelial signalling centres is responsive to Wnt-induced ectodysplasin from the nearby ectoderm [32]. As an inhibitor of Wnt signalling, Dkk4 is a direct transcriptional target of EDA/EDAR signal during lamina formation [80]. Lef-1 is known to play a role in Wnt signalling and transcriptional activation. Recent studies showed that overexpression of both Lef-1 and β-catenin significantly increased EDA transcription, and indirect stabilization of endogenous β-catenin stimulated EDA transcription [48, 70]. The WNT10A protein is a member of the Wnt ligand family [81], which activates the canonical Wnt/β-catenin signalling pathway [21, 82, 83]. In this study, compared with the younger brother, the elder brother exhibited a clinical phenotype with more missing teeth. We speculate that the additional WNT10A variations decreased the binding of mutant WNT10A to Wnt receptor genes (FZD5), resulting in the simultaneous damage of the NF-κB and Wnt signalling pathways and the failure of tooth morphogenesis.
In conclusion, compound heterozygous WNT10A missense variations may exacerbated the number of missing teeth in HED cause by EDA variation. Further study is necessary to determine on how WNT10A interacts with EDA during tooth development.
Availability of data and materials
The datasets analysed during the current study are available in the ClinVar repository (https://www.ncbi.nlm.nih.gov/clinvar/submitters/509028/). The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Cobourne MT, Sharpe PT. Diseases of the tooth: the genetic and molecular basis of inherited anomalies affecting the dentition. Wiley Interdiscip Rev –Develop Biol. 2013;2(2):183–212.
Chhabra N, Goswami M, Chhabra A. Genetic basis of dental agenesis - molecular genetics patterning clinical dentistry. Med Oral Patol Oral Y Cirugia Bucal. 2014;19(2):E112–9.
Salama FS, Abdel-Megid FY. Hypodontia of primary and permanent teeth in a sample of Saudi children. Egyptian dental j. 1994;40(1):625–32.
De Coster PJ, Marks LA, Martens LC, Huysseune A. Dental agenesis: genetic and clinical perspectives. J Oral Pathol Med. 2009;38(1):1–17.
Zhang J, Liu HC, Lyu X, Shen GH, Deng XX, Li WR, Zhang XX, Feng HL. Prevalence of tooth agenesis in adolescent Chinese populations with or without orthodontics. Chin j dental res: the off j Sci Section Chin Stomatol Assoc (CSA). 2015;18(1):59–65.
Stockton DW, Das P, Goldenberg M, D'Souza RN, Patel PI. Mutation of PAX9 is associated with oligodontia. Nat Genet. 2000;24(1):18–9.
Vastardis H, Karimbux N, Guthua SW, Seidman JG, Seidman CE. A human MSX1 homeodomain missense mutation causes selective tooth agenesis. Nat Genet. 1996;13(4):417–21.
Lammi L, Arte S, Somer M, Jarvinen H, Lahermo P, Thesleff I, Pirinen S, Nieminen P. Mutations in AXIN2 cause familial tooth agenesis and predispose to colorectal cancer. Am J Hum Genet. 2004;74(5):1043–50.
Kantaputra P, Sripathomsawat W. WNT10A and isolated Hypodontia. Am J Med Genet A. 2011;155A(5):1119–22.
Yu P, Yang WL, Han D, Wang X, Guo S, Li JC, Li F, Zhang XX, Wong SW, Bai BJ, et al. Mutations in WNT10B are identified in individuals with Oligodontia. Am J Hum Genet. 2016;99(1):195–201.
Massink MPG, Creton MA, Spanevello F, Fennis WMM, Cune MS, Savelberg SMC, Nijman IJ, Maurice MM, van den Boogaard MJH, van Haaften G. Loss-of-function mutations in the WNT co-receptor LRP6 cause autosomal-dominant Oligodontia. Am J Hum Genet. 2015;97(4):621–6.
Song S, Han D, Qu H, Gong Y, Wu H, Zhang X, Zhong N, Feng H. EDA gene mutations underlie non-syndromic Oligodontia. J Dent Res. 2009;88(2):126–31.
van den Boogaard MJ, Creton M, Bronkhorst Y, van der Hout A, Hennekam E, Lindhout D, Cune M, van Amstel HKP. Mutations in WNT10A are present in more than half of isolated hypodontia cases. J Med Genet. 2012;49(5):327–31.
Song SJ, Zhao RY, He HY, Zhang J, Feng HL, Lin LY. WNT10A variants are associated with non-syndromic tooth agenesis in the general population. Hum Genet. 2014;133(1):117–24.
Mikkola ML. Molecular aspects of Hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A(9):2031–6.
Visinoni AF, Lisboa-Costa T, Pagnan NAB, Chautard-Freire-Maia EA. Ectodermal Dysplasias: clinical and molecular review. Am J Med Genet A. 2009;149A(9):1980–2002.
Mikkola ML, Thesleff I. Ectodysplasin signaling in development. Cytokine Growth Factor Rev. 2003;14(3–4):211–24.
Cluzeau C, Hadj-Rabia S, Jambou M, Mansour S, Guigue P, Masmoudi S, Bal E, Chassaing N, Vincent M-C, Viot G, et al. Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of Hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mutat. 2011;32(1):70–7.
Naqvi SK, Wasif N, Javaid H, Ahmad W. Two novel mutations in the gene EDAR causing autosomal recessive hypohidrotic ectodermal dysplasia. Orthodont Craniofac Res. 2011;14(3):156–9.
de Aquino SN, Paranaiba LMR, Swerts MSO, Martelli DRB, de Barros LM, Martelli JH. Orofacial features of hypohidrotic ectodermal dysplasia. Head neck pathol. 2012;6(4):460–6.
Adaimy L, Chouery E, Megarbane H, Mroueh S, Delague V, Nicolas E, Belguith H, de Mazancourt P, Megarbane A. Mutation in WNT10A is associated with an autosomal recessive ectodermal dysplasia: the odonto-onycho-dermal dysplasia. Am J Hum Genet. 2007;81(4):821–8.
Bohring A, Stamm T, Spaich C, Haase C, Spree K, Hehr U, Hoffmann M, Ledig S, Sel S, Wieacker P, et al. WNT10A mutations are a frequent cause of a broad Spectrum of ectodermal Dysplasias with sex-biased manifestation pattern in heterozygotes. Am J Hum Genet. 2009;85(1):97–105.
Zeng B, Xiao X, Li S, Lu H, Lu J, Zhu L, Yu D, Zhao W. Eight mutations of three genes (EDA, EDAR, and WNT10A) identified in seven Hypohidrotic ectodermal dysplasia patients. Genes. 2016;7(9):65.
Chear CT, El Farran BAK, Sham M, Ramalingam K, Noh LM, Ismail IH, Chiow MY, Baharin MF, Ripen AM, Bin MS. A novel De novo NFKBIA missense mutation associated to ectodermal dysplasia with Dysgammaglobulinemia. Genes. 2022;13(10):1900.
Granados EL, Keenan JE, Kinney MC, Leo H, Jain N, Ma CA, Quinones R, Gelfand EW, Jain A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum Mutat. 2008;29(6):861–8.
Courtois G, Smahi A, Reichenbach J, Doffinger R, Cancrini C, Bonnet M, Puel A, Chable-Bessia C, Yamaoka S, Feinberg J, et al. A hypermorphic I kappa B alpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J Clin Investig. 2003;112(7):1108–15.
Jain A, Ma CA, Liu SY, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2(3):223–8.
Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, Shapira SK, Farndon PA, Wara DW, Emmal SA, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet. 2000;67(6):1555–62.
Keller MD, Petersen M, Ong P, Church J, Risma K, Burham J, Jain A, Stiehm ER, Hanson EP, Uzel G, et al. Hypohidrotic ectodermal dysplasia and immunodeficiency with coincident NEMO and EDA mutations. Front Immunol. 2011;2:61.
Yao S, Zhou X, Gu M, Zhang C, Bartsch O, Vona B, Fan L, Ma L, Pan Y. FGFR1 variants contributed to families with tooth agenesis. Human genom. 2023;17(1):93.
Wang HJ, Liu Y, Zheng YF, Zhao XX, Lin SY, Zhang Q, Zhang XY. A novel missense mutation of LRP6 identified by whole-exome sequencing in a Chinese family with non-syndromic tooth agenesis. Orthodont Craniofac Res. 2021;24(2):233–40.
Agostino M, Pohl SO-G. Wnt binding affinity prediction for putative frizzled-type cysteine-rich domains. Int J Mol Sci. 2019;20(17):4168.
Voloshanenko O, Gmach P, Winter J, Kranz D, Boutros M. Mapping of Wnt-frizzled interactions by multiplex CRISPR targeting of receptor gene families. FASEB J. 2017;31(11):4832–44.
Yuan Q, Zhao M, Tandon B, Maili L, Liu X, Zhang A, Baugh EH, Tran T, Silva RM, Hecht JT, et al. Role of WNT10A in failure of tooth development in humans and zebrafish. Molecular Genet Genom Med. 2017;5(6):730–41.
Mustonen T, Ilmonen M, Pummila M, Kangas AT, Laurikkala J, Jaatinen R, Pispa J, Gaide O, Schneider P, Thesleff I, et al. Ectodysplasin A1 promotes placodal cell fate during early morphogenesis of ectodermal appendages. Develop. 2004;131(20):4907–19.
Han D, Gong Y, Wu H, Zhang X, Yan M, Wang X, Qu H, Feng H, Song S. Novel EDA mutation resulting in X-linked non-syndromic hypodontia and the pattern of EDA-associated isolated tooth agenesis. Eur J Med Genet. 2008;51(6):536–46.
Srivastava AK, Pispa J, Hartung AJ, Du Y, Ezer S, Jenks T, Shimada T, Pekkanen M, Mikkola ML, Ko MS, et al. The Tabby phenotype is caused by mutation in a mouse homologue of the EDA gene that reveals novel mouse and human exons and encodes a protein (ectodysplasin-a) with collagenous domains. Proc Natl Acad Sci U S A. 1997;94(24):13069–74.
Yan M, Wang LC, Hymowitz SG, Schilbach S, Lee J, Goddard A, de Vos AM, Gao WQ, Dixit VM. Two-amino acid molecular switch in an epithelial morphogen that regulates binding to two distinct receptors. Science. 2000;290(5491):523–7.
Mikkola ML. TNF superfamily in skin appendage development. Cytokine Growth Factor Rev. 2008;19(3–4):219–30.
Ezer S, Bayés M, Elomaa O, Schlessinger D, Kere J. Ectodysplasin is a collagenous trimeric type II membrane protein with a tumor necrosis factor-like domain and co-localizes with cytoskeletal structures at lateral and apical surfaces of cells. Hum Mol Genet. 1999;8(11):2079–86.
Cui CY, Schlessinger D. EDA signaling and skin appendage development. Cell Cycle. 2006;5(21):2477–83.
Schneider P, Street SL, Gaide O, Hertig S, Tardivel A, Tschopp J, Runkel L, Alevizopoulos K, Ferguson BM, Zonana J. Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-a. J Biol Chem. 2001;276(22):18819–27.
Huang SX, Liang JL, Sui WG, Lin H, Xue W, Chen JJ, Zhang Y, Gong WW, Dai Y, Ou ML. EDA mutation as a cause of hypohidrotic ectodermal dysplasia: a case report and review of the literature. Genet Mol Res. 2015;14(3):10344–51.
He F, Wang H, Zhang X, Gao Q, Guo F, Chen C. Conservation analysis and pathogenicity prediction of mutant genes of ectodysplasin a. Bmc Med Genet. 2018;19(1):209.
Huxford T, Ghosh G. A structural guide to proteins of the NF-kappaB signaling module. Cold Spring Harb Perspect Biol. 2009;1(3):a000075.
Kanarek N, Ben-Neriah Y. Regulation of NF-κB by ubiquitination and degradation of the IκBs. Immunol Rev. 2012;246(1):77–94.
Reyes-Reali J, Isabel Mendoza-Ramos M, Garrido-Guerrero E, Mendez-Catala CF, Mendez-Cruz AR, Pozo-Molina G. Hypohidrotic ectodermal dysplasia: clinical and molecular review. Int J Dermatol. 2018;57(8):965–72.
Kowalczyk-Quintas C, Schneider P. Ectodysplasin a (EDA) - EDA receptor signalling and its pharmacological modulation. Cytokine Growth Factor Rev. 2014;25(2):195–203.
Liu YS, Huang YZ, Hua R, Zhao XL, Yang W, Liu YP, Zhang X. Mutation screening of the EDA gene in seven Chinese families with X-linked Hypohidrotic ectodermal dysplasia. Genetic Test Molecular Biomark. 2018;22(8):487–91.
Liu XY, Zhu JX, Zhao YM. Whole exome sequencing and analysis of hypohidrotic ectodermal dysplasia patients. Zhonghua kou qiang yi xue za zhi = Zhonghua kouqiang yixue zazhi =. Chin j stomatol. 2022;57(2):155–61.
Wang X, Zhang ZY, Yuan S, Ren JB, Qu H, Zhang GZ, Chen WJ, Zheng SS, Meng LQ, Bai JP, et al. A novel EDA1 missense mutation in X-linked hypohidrotic ectodermal dysplasia. Medicine. 2020;99(11):e19244.
Savasta S, Carlone G, Castagnoli R, Chiappe F, Bassanese F, Piras R, Salpietro V, Brazzelli V, Verrotti A, Marseglia GL. X-linked Hypohidrotic ectodermal dysplasia: new features and a novel EDA gene mutation. Cytogenet Genome Res. 2017;152(3):111–6.
Barbato E, Traversa A, Guarnieri R, Giovannetti A, Genovesi ML, Magliozzi MR, Paolacci S, Ciolfi A, Pizzi S, Di Giorgio R, et al. Whole exome sequencing in an Italian family with isolated maxillary canine agenesis and canine eruption anomalies. Arch Oral Biol. 2018;91:96–102.
Wu JY, Yu M, Sun SC, Fan ZZ, Zheng JL, Zhang LT, Feng HL, Liu Y, Han D. Detection of EDA gene mutation and phenotypic analysis in patients with hypohidrotic ectodermal dysplasia. Beijing Da Xue Xue Bao. 2020;53(1):24–33.
Park JS, Ko JM, Chae JH. Novel and private EDA mutations and clinical phenotypes of Korean patients with X-linked Hypohidrotic ectodermal dysplasia. Cytogenet Genome Res. 2019;158(1):1–9.
Alksere B, Kornejeva L, Grinfelde I, Dzalbs A, Enkure D, Conka U, Andersone S, Blumberga A, Nikitina-Zake L, Kangare L, et al. A novel EDA variant causing X-linked hypohidrotic ectodermal dysplasia: case report. Molecular Genet Metab Rep. 2021;29:100796.
Shen L, Liu CY, Gao M, Li HM, Zhang YW, Tian Q, Ni HL, Peng PW, Zhao RJ, Hu ZM, et al. Novel mutation of EDA causes new asymmetrical X-linked hypohidrotic ectodermal dysplasia phenotypes in a female. J Dermatol. 2019;46(8):731–3.
Zhao K, Lian MF, Zou DH, Huang W, Zhou WJ, Shen YH, Wang F, Wu YQ. Novel mutations identified in patients with tooth agenesis by whole-exome sequencing. Oral Dis. 2019;25(2):523–34.
Zeng BH, Zhao Q, Li SJ, Lu H, Lu JX, Ma L, Zhao W, Yu DS. Novel EDA or EDAR mutations identified in patients with X-linked Hypohidrotic ectodermal dysplasia or non-syndromic tooth agenesis. Genes. 2017;8(10):259.
Parveen A, Khan SA, Mirza MU, Bashir H, Arshad F, Iqbal M, Ahmad W, Wahab A, Fiaz A, Naz S, et al. Deleterious variants in WNT10A, EDAR, and EDA causing isolated and syndromic tooth agenesis: a structural perspective from molecular dynamics simulations. Int J Mol Sci. 2019;20(21):5282.
Okubo A, Fujii K, Arimura A, Katsue H, Higashi Y, Shimomura Y, Kanekura T. Hypohidrotic ectodermal dysplasia with strabismus. J Dermatol. 2018;45(7):E191–2.
Xu XG, Lv Y, Yan HW, Qu L, Xiao T, Geng L, He CD, Liu CX, Gao XH, Li YH, et al. Next-generation sequencing identified a novel EDA mutation in a Chinese pedigree of Hypohidrotic ectodermal dysplasia with hyperplasia of the sebaceous glands. Acta Derm Venereol. 2017;97(8):984–5.
Andreoni F, Sgattoni C, Bencardino D, Simonetti O, Forabosco A, Magnani M. Missense mutations in EDA and EDAR genes cause dominant syndromic tooth agenesis. Molecular Genet Gen Med. 2021;9(1):e1555.
Liu GN, Wang X, Qin M, Sun LS, Zhu JX. A novel missense mutation p.S305R of EDA gene causes XLHED in a Chinese family. Arch Oral Biol. 2019;107:104507.
Nakajima M, Hayashi R, Shinkuma S, Watanabe M, Shigehara Y, Shimomura Y, Abe R. Two cases of hypohidrotic ectodermal dysplasia caused by novel deletion mutations in the EDA gene. J Dermatol. 2019;46(1):E21–2.
Mintoff D, Pace NP, Mercieca V, Bauer P, Borg I. A novel c.916C > a EDA gene pathogenic variant in a boy with X-linked hypohidrotic ectodermal dysplasia. Clin Exp Dermatol. 2021;46(3):618–20.
Rahbaran M, Doabsari MH, Salavitabar S, Mokhberian N, Morovvati Z, Morovvati S. A novel frameshift mutation in the EDA gene in an Iranian patient affected by X-linked hypohidrotic ectodermal dysplasia. Cell Molecular Biol Lett. 2019;24:54.
Miyake T, Kiniwa Y, Kosho T, Nakano H, Okuyama R. Hypohidrotic ectodermal dysplasia: a report of two cases. J Dermatol. 2017;44(4):479–81.
Lei K, Zhang Y, Dong Z, Sun Y, Yi Z, Chen Z. A novel 1-bp deletion mutation and extremely skewed X-chromosome inactivation causing severe X-linked hypohidrotic ectodermal dysplasia in a Chinese girl. Clin Exp Dermatol. 2018;43(1):60–2.
Yu M, Wong S-W, Han D, Cai T. Genetic analysis: Wnt and other pathways in nonsyndromic tooth agenesis. Oral Dis. 2019;25(3):646–51.
Yu M, Liu Y, Liu H, Wong S-W, He H, Zhang X, Wang Y, Han D, Feng H. Distinct impacts of bi-allelic WNT10A mutations on the permanent and primary dentitions in odonto-onycho-dermal dysplasia. Am J Med Genet A. 2019;179(1):57–64.
Garcia-Martinez V-E, Galiana-Valles X, Zomeno-Alcala O, Rodriguez-Lopez R, Llena C, Martinez-Romero MC, Guillen-Navarro E. Dental phenotype with minor ectodermal symptoms suggestive of WNT10A deficiency. Children-Basel. 2023;10(2):356.
Kanchanasevee C, Sriwattanapong K, Theerapanon T, Thaweesapphithak S, Chetruengchai W, Porntaveetus T, Shotelersuk V. Phenotypic and genotypic features of Thai patients with nonsyndromic tooth agenesis and WNT10A variants. Front Physiol. 2020;11:573214.
Machida J, Goto H, Tatematsu T, Shibata A, Miyachi H, Takahashi K, Izumi H, Nakayama A, Shimozato K, Tokita Y. WNT10A variants isolated from Japanese patients with congenital tooth agenesis. Human genome variat. 2017;4:17047.
Liu HC, Lin BC, Liu HB, Su LX, Feng HL, Liu Y, Yu M, Han D. Dose dependence effect in Biallelic WNT10A variant-associated tooth agenesis phenotype. Diagnostics. 2022;12(12):3087.
Zeng Y, Baugh E, Akyalcin S, Letra A. Functional effects of WNT10A rare variants associated with tooth agenesis. J Dent Res. 2021;100(3):302–9.
Kantaputra P, Kaewgahya M, Jotikasthira D, Kantaputra W. Tricho- Odonto- Onycho- dermal dysplasia and WNT10A mutations. Am J Med Genet A. 2014;164(4):1041–8.
He H, Han D, Feng H, Qu H, Song S, Bai B, Zhang Z. Involvement of and interaction between WNT10A and EDA mutations in tooth agenesis cases in the Chinese population. PLoS One. 2013;8(11):e80393.
Zhang Y, Tomann P, Andl T, Gallant NM, Huelsken J, Jerchow B, Birchmeier W, Paus R, Piccolo S, Mikkola ML, et al. Reciprocal requirements for EDA/EDAR/NF-kappaB and Wnt/beta-catenin signaling pathways in hair follicle induction. Dev Cell. 2009;17(1):49–61.
Cui CY, Kunisada M, Piao Y, Childress V, Ko MS, Schlessinger D. Dkk4 and Eda regulate distinctive developmental mechanisms for subtypes of mouse hair. PLoS One. 2010;5(4):e10009.
Bielik P, Bonczek O, Krejci P, Zeman T, Izakovicova-Holla L, Soukalova J, Vanek J, Vojtesek B, Lochman J, Balcar VJ, et al. WNT10A variants: following the pattern of inheritance in tooth agenesis and self-reported family history of cancer. Clin Oral Investig. 2022;26(12):7045–55.
Yin W, Bian Z. The gene network underlying Hypodontia. J Dent Res. 2015;94(7):878–85.
Williams MA, Letra A. The changing landscape in the genetic etiology of human tooth agenesis. Genes. 2018;9(5):255.
Acknowledgements
We thank the patients for their generous contribution to this work.
Web resources
1000 genomes: https://www.internationalgenome.org/.
Exome Aggregation Consortium: http://exac.broadinstitute.org/.
Genome Aggregation Database: https://gnomad.broadinstitute.org/.
UCSC Genome Browser: http://genome.ucsc.edu/.
NCBI: https://www.ncbi.nlm.nih.gov/tools/primer-blast/.
HDOCK: http://hdock.phys.hust.edu.cn/.
PsiPred 4.0: http://bioinf.cs.ucl.ac.uk/psipred/.
AlphaFold 2: https://www.alphafold.ebi.ac.uk/.
Funding
This work was supported by National Natural Science Foundation of China (grant no. 82022014), the Key Research and Development Program of Science and Technology Department of Hunan Province, China (grant no. 2022SK2049) and Changsha Municipal Natural Science Foundation (grant no. kq2208394).
Author information
Authors and Affiliations
Contributions
The first draft of the manuscript was written by Yiting Liu, Jing Sun, and Caiqi Zhang. Qingping Gao and Xiaoshan Wu supervised the study and edited the manuscript. Siyuan Ma, Yi Wu and Xuechun Li performed data analysis. All authors commented on previous versions of the manuscript and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Conducted the study in accordance with the Declaration of Helsinki. All procedures performed in studies were approved by the Medical Ethic Committee of Xiangya Hospital of Central South University (Approval Number: 202203062). Informed consent was obtained from all individual participants included in the study. As the participants were under the age of 16, the informed consent was fully obtained from the participants’ parents or legal guardians.
Consent for publication
Informed consent was obtained from the parents/legal guardians of the participants for publication of identifying images and information.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liu, Y., Sun, J., Zhang, C. et al. Compound heterozygous WNT10A missense variations exacerbated the tooth agenesis caused by hypohidrotic ectodermal dysplasia. BMC Oral Health 24, 136 (2024). https://doi.org/10.1186/s12903-024-03888-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12903-024-03888-5