
Abstract
The environment of healthcare institutes (HCIs) potentially affects the internal microecology of medical workers, which is reflected not only in the well-studied gut microbiome but also in the more susceptible oral microbiome. We conducted a prospective cross-sectional cohort study in four hospital departments in Central China. Oropharyngeal swabs from 65 healthcare workers were collected and analyzed using 16S rRNA gene amplicon sequencing. The oral microbiome of healthcare workers exhibited prominent deviations in diversity, microbial structure, and predicted function. The coronary care unit (CCU) samples exhibited robust features and stability, with significantly higher abundances of genera such as Haemophilus, Fusobacterium, and Streptococcus, and a lower abundance of Prevotella. Functional prediction analysis showed that vitamin, nucleotide, and amino acid metabolisms were significantly different among the four departments. The CCU group was at a potential risk of developing periodontal disease owing to the increased abundance of F. nucleatum. Additionally, oral microbial diversification of healthcare workers was related to seniority. We described the oral microbiome profile of healthcare workers in different clinical scenarios and demonstrated that community diversity, structure, and potential functions differed markedly among departments. Intense modulation of the oral microbiome of healthcare workers occurs because of their original departments, especially in the CCU.
Similar content being viewed by others

Introduction
Unique conditions render the microbial composition in healthcare institutes (HCIs) vastly different from that in the external natural environment [1,2,3]. Hospitalized individuals are at a greater risk of human-related microorganisms or pathogens colonizing their nasal cavity compared with non-hospitalized groups [4]. Additionally, the diversity and compositions of the microbiome change dynamically in different clinical scenarios, which may be correlated with different indoor environmental conditions [5]. A previous study reported that the gut microbiome of intensive care unit (ICU) workers, compared with non-ICU workers, showed a significantly increased abundance of Dialister, Enterobacteriaceae, Phascolarctobacterium, Pseudomonas, Veillonella, and Streptococcus and a marked depletion of Faecalibacterium, Blautia, and Coprococcus [6]. Beyond the aspect of common colonizing microorganisms, potential pathogens in hospital settings cannot be neglected. Thus, different clinical scenarios in HCIs may affect the microbiota of healthcare workers. Microbial ecology in HCIs comprehensively affects the health of healthcare workers. Studies have shown that the adverse microbial characteristics of HCIs could increase the incidence of microbial infections in staff [7,8,9]. Therefore, identifying the microbial status of healthcare workers is of extreme importance in the control of nosocomial infections.
Previous studies have widely focused on stool samples from healthcare workers to elucidate gut microbial properties, while respiratory samples are rarely involved. The respiratory tract is a common route of nosocomial infections [10], and the occurrence and progression of many diseases have been related to alterations in the respiratory microbiome [11,12,13]. The oral microbiome closely resembles that of the lung [14]. Numerous empirical studies have illustrated the dominance of Firmicutes, Actinobacteria, Proteobacteria, Fusobacteria, Bacteroidetes, and Spirochaetes in a healthy oral cavity, constituting 96% of the total oral bacteria [15, 16]. It has been noted that specific genera or species play roles in oral health and disease, even extraoral sites in systemic diseases [17]. Hence, exploring the extent of the oral microbiome through non-invasive operations may reflect more internal respiratory tract characterizations [18, 19]. Meanwhile, 16 S rRNA gene sequences have potential advantages for detecting the oral microbiome, whether they are cultivated or not [17].
Currently, the composition of the oral microbiome of healthcare workers and the influence of the HCI environment remain unclear. Here, we profiled the microbial community of oropharyngeal swabs from healthcare workers in different clinical scenarios in a hospital based on 16 S rRNA gene sequencing targeting multiple bacterial hypervariable regions. Oral microbial composition and predicted functional characteristics were analyzed to evaluate the impact of the hospital environment on the oral microbiome of healthcare workers.
Methods
Participants and sample collection
A total of 65 full-time healthcare workers from coronary care unit (CCU, n = 12), ICU (n = 16), operating room (OR, n = 16) and department of respiratory medicine (RES, n = 21) of Linfen Central Hospital (Shanxi Province, China) were recruited. Subjects were excluded if they had a respiratory tract infection or respiratory tract disease, or were treated with antibiotics in three months prior to sampling, or had worked less than one year in the hospital. Participants were asked to avoid eating and drinking for three hours prior to sampling. All fresh oropharyngeal swabs were collected by one operator within two hours and immediately stored at −80 °C until DNA extraction. Additional information on age, gender, position, seniority, sleeping and dietary habits were obtained through questionnaires.
DNA preparation and sequencing
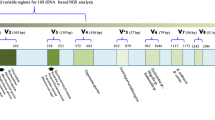
Total genomic DNA was extracted from oropharyngeal swabs using a TIANamp Micro DNA Kit (Tiangen, China) following the manufacturer’s instructions. We amplified the corresponding hypervariable regions (V2, V3, V4, V6-7, V8 and V9) of the 16 S rRNA with two primer pools in an Ion 16 S™ Metagenomics Kit (ThermoFisher Scientific, UK). A total of six primer pairs amplifying multiple hypervariable regions listed above were split into two pools to avoid possible interference during the amplification reaction. Every DNA template was amplified in two primer pools, and then two tubes of PCR products were combined to obtain complete amplification products from multiple hypervariable regions. XP beads were next used to purify the amplification products and quantified by Qubit4 (ThermoFisher Scientific, USA). Purified amplicons were ligated with barcodes to prepare the libraries. Then, libraries were pooled in equimolar amounts on chip 530 and sequenced to single-end, 250-base-pair reads on an Ion GeneStudio S5 System (ThermoFisher Scientific, USA) based on the Ion Reporter metagenomics workflow (Ion 16 S mNGS), which had 100% sensitivity when accounting for the genus level of detection [20]. All amplified regions were sequenced. Sequencing of multiple variable regions allows for higher resolution.
Bioinformatic analyses
Quality filtering, trimming and dereplication of raw sequencing reads were conducted automatically on the Ion Reporter metagenomics workflow, relying on default parameters. We next used the UCHIME algorithm [21] to remove the chimeric sequences and used unoise3 [22] to generate denoising amplicon sequence variants (ASVs). Taxonomy assignment was performed based on vsearch [23] referring to the SILVA (V 138.1) [24] and GreenGene database [25] with a threshold of 97%. Reads with an alignment rate below 97% were rejected. We aligned reads from different variable regions with the bacterial reference genome separately. A consensus table was created by summing all read counts from different regions with identical taxonomic rank detection. We rarefied the sequencing data and then evaluated the alpha diversity by the Good’s average index, Chao1 index, abundance-based coverage estimator (ACE) index, Shannon index and Simpson index. Permutational multivariate analysis of variance (PERMANOVA) and analysis of similarity (ANOSIM) based on the Bray‒Curtis distance were used to evaluate the beta diversity.
Differential bacterial taxa among groups were obtained using linear discriminant analysis effect size (LEfSe) [26] with the criteria of LDA > 2 (or LDA > 4) and P < 0.05. Microbiome phenotypes were predicted using BugBase [27]. PICRUSt2 [28] was used to identify microbiome-associated pathways from the inferred metagenomes of taxa using the ‘stratified’ option. We applied the Pearson correlation algorithm to identify associations across bacterial genera, representing correlation strength and assigned them to the edges. For the Pearson correlation table, we used the cytoHubba [29] plugin in Cytoscape [30] (V 3.9.1) to find Hubba nodes based on the maximum cross-correlation algorithm. The Hubba nodes represent the taxonomy that had the highest correlation with the other genera. Then, we took the intersection of correlation nodes and the top 10 Hubba nodes and retained nodes whose absolute correlation value was greater than 0.6.
Statistical analysis
Parametric continuous variables are presented as mean ± standard deviation, and abnormally distributed continuous variables are presented as medians and interquartile range (25th and 75th percentiles). Categorical variables are described as numbers (percentages). Student’s t-test and analysis of variance (ANOVA) with post hoc Tukey HSD test were used to compare continuous parametric data conforming to normal distribution. Abnormally distributed continuous variables were compared using the Mann‒Whitney U test or Kruskal‒Wallis H-test. Categorical variables were analyzed using the chi-square or Fisher’s test. All tests were two-sided. P values were corrected using FDR, and P < 0.05 was considered statistically significant [31]. We performed statistical analyses by using SPSS version 25 software.
Results
Demographic characteristics of the study population
We investigated the demographics of the participants, including several indicators that may affect the oral microbiome [32, 33]. The statistical results are presented in Table 1. The mean age was 32.03 years old, and 76.9% were female. A total of 58.5% of the participants were nursing staff, and the remainder were resident doctors. The mean seniority was 7.86 years. The median number of sleeping hours per day and intake times of sweets/desserts per week were 6.50 and 3.00, respectively. There were no significant differences in these parameters among the CCU, ICU, OR, and RES groups. All participants were free of metabolic diseases such as diabetes and hyperlipidemia.
Distinct oral microbial structure and diversity among departments
The rarefaction curves of all samples demonstrated that the sequencing coverage was sufficient to represent the microbial composition (Supplementary Material Fig. S1). A total of 13,251 ASVs were identified in all 65 samples. After filtering out ASVs found in less than two samples, 1519 ASVs were reserved for downstream analysis with a minimum relative abundance of 0.05% [34].
We first assessed alpha diversity within individuals. The CCU group showed the lowest level of microbial diversity compared to the other groups, which was reflected in the Shannon and Good’s coverage index (Fig. 1A, Supplementary Material Fig. S2). In terms of beta diversity, PERMANOVA based on Bray–Curtis distance hardly exhibited obvious intergroup clustering of microbiota structure, even if statistically significant (Fig. 1B). Furthermore, ANOSIM analysis for pairwise comparison showed that the bacterial community of the CCU provided the most significant between-group differential components (Supplementary Material Fig. S3). We also tested whether the oral microbiome of healthcare workers is associated with other demographic characteristics. The department proved to be the main grouping factor accounting for the variance of the oral microbiota compared to other confounders, such as age, sex, position, diet, and sleep, although there was also a significant difference between those with 5–10 years of seniority and those with more than 10 years of seniority (Supplementary Material Fig. S3, P = 0.027). Taken together, these findings revealed prominent differences in oral microbial structure and diversity among the departments.

Structure and alteration of the oral microbiome in healthcare workers from different departments
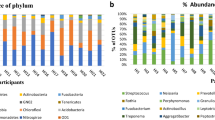
(A) Alpha diversity indices, including the Shannon index (left, ANOVA test, CCU versus ICU P = 0.007, CCU versus RES P = 0.007) and Good’s average index (right, Kruskal‒Wallis test, CCU versus ICU P = 0.000, CCU versus OR P = 0.005, ICU versus RES P = 0.000, OR versus RES P = 0.011), represent differences in within-sample diversity among departments. (B) PCoA plot showing significant deviation among departments based on PERMANOVA at the genus level. Box plots of the primary (down) and second (left) constrained axes are shown. (C) Stacked bar chart showing the relative abundance of departments at the phylum level. (D) Box plot revealing significant deviation in the relative abundance of Spirochaetes. * P < 0.05; ** P < 0.01; *** P < 0.001
Differences in the oral microbiome between hospital departments
The microbiome profile comprised 13 phyla, 20 classes, 29 orders, 53 families, and 97 genera. Core phyla were defined as those identified in all samples. Nine core phyla, Bacteroidetes, Firmicutes, Proteobacteria, Actinobacteria, Fusobacteria, Candidatus Saccharibacteria, Spirochaetes, SR1, and Synergistetes, are shown in Fig. 1C. We conducted the Kruskal–Wallis test to compare the relative abundance of the nine core phyla across groups, and Spirochaetes was significant (Fig. 1D, CCU versus ICU P = 0.000; CCU versus RES P = 0.001; ICU versus OR P = 0.005; OR versus RES P = 0.043). We further conducted LEfSe analysis to identify significant differences in abundance between departments, considering CCU as the basic group. We identified 66 microbial taxa (19 CCU-enriched versus 47 ICU-enriched) that differed significantly in relative abundance between CCU and ICU, 36 microbial taxa (14 CCU-enriched versus 22 OR-enriched) that differed significantly in relative abundance between CCU and OR, and 40 microbial taxa (10 CCU-enriched versus 30 RES-enriched) that differed significantly in relative abundance between CCU and RES (Supplementary Table S1). To identify more reliable differential taxa, we set a stricter filter with LDA > 4. The relative abundance of Haemophilus in the CCU group was higher than that in the other groups, while Prevotella showed the opposite trend. Moreover, compared with ICU and RES workers, CCU workers showed a higher abundance of taxa belonging to Fusobacteria (Fusobacterium) and Firmicutes (Streptococcus). The relative abundance of Bacilli, a class belonging to Firmicutes, in the CCU suggested a level of depletion to the OR (Fig. 2A-C, Supplementary Material Fig. S4).

Department-associated microbial taxa identified via LEfSe analysis
Cladogram plots showing the significant differences in relative abundance (LDA > 4) between ICU (A), RES (B) and OR (C) versus CCU. (D) Co-occurrence networks showing associations among significant oral microbial taxa based on Pearson correlation and maximum cross correlation algorithm. The size of the nodes indicates their degree of hub. Connecting lines represent the Pearson correlation coefficient ρ > 0.6 (red line) or < -0.6 (green line). (E) Box plot showing the relative abundance of F. nucleatum (CCU versus OR, P = 0.002) and P. gingivalis (CCU versus ICU, P = 0.04; OR versus ICU, P = 0.016) across departments. The Kruskal‒Wallis test was used
We further evaluated the effects of various grouping characteristics on the oral microbiome. Factors such as age, sex, position, diet, and sleep could not be used to distinguish the differential microbiota. Notably, when we grouped the participants by seniority, several differential bacterial genera were identified (Supplementary Material Fig. S5), suggesting that oral microbial diversification of healthcare workers is related to seniority.
We next performed co-occurrence network analysis and found vital interconnections within the oral microbiome, indicating that these healthcare-worker-altered taxa did not occur independently in the oral environment. Megasphaera, Prevotella, Leptotrichia, Atopobium, and Veillonella may be essential genera that shape the oral microbiome of healthcare workers, accompanied by rich multivariate interrelationships and strong correlations between each other (Fig. 2D). Porphyromonas was negatively correlated with Prevotella, whereas the rest were positively correlated.
Differences in periodontal pathogens
We further explored the distribution of critical periodontal pathogens among departments [35,36,37,38,39,40]. The relative abundance of F. nucleatum increased significantly in the CCU group compared to the OR group, but compared to the ICU group the increase was not significant (Fig. 2E). The relative abundance of P. gingivalis in the ICU was higher than that in the OR or CCU. However, the relative abundance of P. gingivalis was approximately 10 times lower than that of F. nucleatum (Supplementary Material Fig. S6). These results suggest a potential risk of periodontal disease in the CCU and the ICU.
Potential function of the oral microbiome
We analyzed the predicted phenotypes based on taxonomic classification using BugBase. BugBase categorized six main bacterial phenotype categories: Gram staining, oxygen tolerance, ability to form biofilms, mobile element content, pathogenicity, and oxidative stress tolerance. Phenotypes were inferred based on experimental data and pathway/gene presence information collected from various databases, such as Integrated Microbial Genomes (IMG) and the PathoSystems Resource Integration Center (PATRIC) [27]. BugBase data between departments were compared using pairwise Mann-Whitney-Wilcoxon test. Facultative anaerobic bacteria were more abundant in the CCU group than in the other groups (CCU versus ICU, P = 0.017; CCU versus OR, P = 0.047; CCU versus RES, P = 0.048; Supplementary Material Fig. S7A). The relative abundance of gram–positive bacteria in the CCU and OR groups was higher than that in the RES group (CCU versus RES, P = 0.000; OR versus RES, P = 0.043), whereas the opposite was true for gram–negative bacteria (CCU versus RES, P = 0.000; OR versus RES, P = 0.043; Supplementary Material Fig. S7B–C).
PICRUSt2 was employed to impute MetaCyc pathway abundance from the original taxonomic assignment. In total, 399 pathways were annotated. Metabolic pathway data were compared by two-sided Welch’s t–test and filtered for false discoveries using the Benjamini-Hochberg method. Items with q-values less than 0.05 were considered significant. Sixteen differential pathways were elucidated between CCU and ICU, two of which were responsible for nucleotide biosynthesis (“pyrimidine deoxyribonucleotides de novo biosynthesis II” and “superpathway of purine nucleotides de novo biosynthesis II”) and were enriched in CCU (Fig. 3A). In addition, vitamin B12 synthesis was also upregulated in CCU(“adenosylcobalamin biosynthesis from cobyrinate a,c-diamide I” and “adenosylcobalamin salvage from cobinamide II”) (Fig. 3A). We identified 15 differential pathways between the CCU and OR groups. Nucleotide and vitamin B12 biosynthesis processes (“superpathway of purine nucleotides de novo biosynthesis II,” “pyrimidine deoxyribonucleotides de novo biosynthesis II,” “adenosylcobalamin biosynthesis from cobyrinate a,c-diamide I,” and “adenosylcobalamin salvage from cobinamide II”) were more active in CCU (Fig. 3B). Finally, the functional catalog, including biosynthesis and degradation of nucleotides, amino acids and starch, appeared to be enriched in the RES group compared to that in the CCU group (Fig. 3C). These results suggest that the predicted microbial functions of vitamin, nucleotide and amino acid metabolism were significantly different between the departments.

Functional characterization of different groups based on PICRUSt2 analysis
Bar chart showing the functional difference (corrected q–value < 0.05) between ICU (A), OR (B) and RES (C) versus CCU. Data were compared by two-sided Welch’s t–test and filtered for false discoveries using the Benjamini-Hochberg method. Items with q-values less than 0.05 are shown in the figure. The above analyses were all performed on stamp software [52].
Discussion
Healthcare workers are exposed to hospital environments and are constantly in contact with infected patients during daily work. High-risk exposure to transmissible bacteria affects not only the microbiome of the skin surface, but also the respiratory and digestive tracts. To the best of our knowledge, no study has adequately described the characteristics of the oral microbiome in healthcare workers. We demonstrated that the bacterial community diversity, structure, and potential function of staff in the CCU, ICU, OR, and RES departments differed markedly.
Since late 2019/early 2020, the COVID-19 pandemic has led to general universal masking in healthcare settings. Our study reflects the differences in the oral microbiota composition of healthcare workers from different HCI environments during the pandemic. We selected a single-center hospital in Shanxi Province, China, for the study, which excluded the influence of diet, the living environment, and cultural background as much as possible [41,42,43]. It can be speculated that the original departments led to the different compositions of the oral microbial community. In the analysis of beta diversity, our data suggested that the CCU contributed the most significant between-group differences. In the subsequent comparison of oral microbial compositional differences and functional analysis, the CCU also exhibited robust features and stability. We inferred that the oral microbiome of the CCU healthcare workers received characteristic modulations from their departments. Thus, it was reasonable to consider the CCU as the basic object for comparison with other groups. Microbial distribution showed deviation among departments, with an increased abundance of Spirochaetes in the ICU and RES. Numerous empirical studies have shown that oral Spirochaetes cause damage to periodontal tissue by the direct effect of bacterial enzymes and cytotoxic products of bacterial metabolism [44,45,46].
It seems that the performance of F. nucleatum was more weighted than that of P. gingivalis because of its greater relative abundance. Taken together, these findings indicate that healthcare workers in different departments face specific risks of periodontal disease.
At the genus level, the CCU group showed significant differences compared to other groups, with an elevated abundance of Haemophilus and decreased abundance of Prevotella, demonstrating a possible impact on the oral microbiome of healthcare workers in different clinical scenarios. In general, exposure to Haemophilus is most common in the department of respiratory medicine [47, 48]. However, the prevalence of Haemophilus spp. in the CCU was higher in our study. It has been reported that Haemophilus accounts for most gram–negative bacilli causing infective endocarditis [49,50,51], which is usually treated in the CCU. In contrast, the relatively depleted abundance of Haemophilus in the RES may represent stricter protection for healthcare workers in the Department of Respiratory Medicine.
This study had several limitations. First, the study design was not longitudinal, and it lacked long-term tracking and analysis. Second, to explore the stabilizing effects, we selected enrollees working for over one year. More studies are needed to monitor oral microbial changes in short-term healthcare workers. Third, we did not clarify how nonbacterial microbiota (fungi, viruses, and archaea) contribute to the oral microbiome. Finally, despite the observation of different oral microbiomes among departments, we could neither decipher the causal relationships of the differences nor evaluate the influence of such differences on the health of the participants.
Conclusion
In this study, we provide a profile of the oral microbiome of healthcare workers and highlight the essential role of the HCI environment. Workers in the CCU are more likely to exhibit inherent microbiological characteristics, such as reduced diversity, significantly differentiated genera, and higher potential for periodontal diseases. Our study provides a reference for further understanding of the oral microbiological characteristics of healthcare workers. In light of our results, we propose that continuous monitoring of the oral microbiome of healthcare workers in different clinical scenarios should be considered to improve health.
Data availability
The datasets generated and analyzed during the current study are available in the NCBI repository (http://www.ncbi.nlm.nih.gov/bioproject/841067, BioProject ID PRJNA841067).
Change history
22 December 2022
A Correction to this paper has been published: https://doi.org/10.1186/s12903-022-02667-4
Abbreviations
- HCI:
-
Healthcare institute
- ASV:
-
Amplicon sequence variant
- CCU:
-
Coronary care unit
- ICU:
-
Intensive care unit
- OR:
-
Operating room
- RES:
-
Department of respiratory medicine.
References
Poza M, Gayoso C, Gómez MJ, et al. Exploring bacterial diversity in hospital environments by GS-FLX Titanium pyrosequencing. PLoS ONE. 2012;7(8):e44105. DOI:https://doi.org/10.1371/journal.pone.0044105. (In eng).
Kelley ST, Gilbert JA. Studying the microbiology of the indoor environment. Genome Biol. 2013;14(2):202. DOI:https://doi.org/10.1186/gb-2013-14-2-202. (In eng).
Lax S, Sangwan N, Smith D, et al. Bacterial colonization and succession in a newly opened hospital. Sci Transl Med 2017;9(391) (In eng). DOI:https://doi.org/10.1126/scitranslmed.aah6500.
Chen CH, Liou ML, Lee CY, Chang MC, Kuo HY, Chang TH. Diversity of nasal microbiota and its interaction with surface microbiota among residents in healthcare institutes. Sci Rep. 2019;9(1):6175. DOI:https://doi.org/10.1038/s41598-019-42548-5. (In eng).
Chen C-H, Lin Y-L, Chen K-H, et al. Bacterial diversity among four healthcare-associated institutes in Taiwan. Sci Rep. 2017;7(1):8230–0. DOI:https://doi.org/10.1038/s41598-017-08679-3. (In eng).
Zheng N, Li SH, Dong B, et al. Comparison of the gut microbiota of short-term and long-term medical workers and non-medical controls: a cross-sectional analysis. Clin Microbiol Infect. 2021;27(9):1285–92. DOI:https://doi.org/10.1016/j.cmi.2020.10.033. (In eng).
Adams CE, Dancer SJ. Dynamic Transmission of Staphylococcus Aureus in the Intensive Care Unit. Int J Environ Res Public Health 2020;17(6) (In eng). DOI:https://doi.org/10.3390/ijerph17062109.
Dahiya S, Chhillar AK, Sharma N, et al. Candida auris and Nosocomial Infection. Curr Drug Targets. 2020;21(4):365–73. DOI:https://doi.org/10.2174/1389450120666190924155631. (In eng).
Wang X, Zhou Q, He Y, et al. Nosocomial outbreak of COVID-19 pneumonia in Wuhan, China. Eur Respir J 2020;55(6) (In eng). DOI:https://doi.org/10.1183/13993003.00544-2020.
Kollef MH, Torres A, Shorr AF, Martin-Loeches I, Micek ST. Nosocomial Infection. Crit Care Med. 2021;49(2):169–87. DOI:https://doi.org/10.1097/ccm.0000000000004783.
Moffatt MF, Cookson WO. The lung microbiome in health and disease. Clin Med (Lond). 2017;17(6):525–9. DOI:https://doi.org/10.7861/clinmedicine.17-6-525. (In eng).
Mammen MJ, Sethi S. COPD and the microbiome. Respirology. 2016;21(4):590–9. DOI:https://doi.org/10.1111/resp.12732. (In eng).
Lanaspa M, Bassat Q, Medeiros MM, Muñoz-Almagro C. Respiratory microbiota and lower respiratory tract disease. Expert Rev Anti Infect Ther. 2017;15(7):703–11. DOI:https://doi.org/10.1080/14787210.2017.1349609. (In eng).
Dickson RP, Erb-Downward JR, Martinez FJ, Huffnagle GB. The Microbiome and the Respiratory Tract. Annu Rev Physiol. 2016;78:481–504. DOI:https://doi.org/10.1146/annurev-physiol-021115-105238. (In eng).
Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259. DOI:https://doi.org/10.1186/1471-2180-9-259. (In eng).
Bik EM, Long CD, Armitage GC, et al. Bacterial diversity in the oral cavity of 10 healthy individuals. Isme j. 2010;4(8):962–74. DOI:https://doi.org/10.1038/ismej.2010.30. (In eng).
Krishnan K, Chen T, Paster BJ. A practical guide to the oral microbiome and its relation to health and disease. Oral Dis. 2017;23(3):276–86. DOI:https://doi.org/10.1111/odi.12509. (In eng).
Venkataraman A, Bassis CM, Beck JM, et al. Application of a neutral community model to assess structuring of the human lung microbiome. mBio 2015;6(1) (In eng). DOI:https://doi.org/10.1128/mBio.02284-14.
Bassis CM, Erb-Downward JR, Dickson RP, et al. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio. 2015;6(2):e00037. DOI:https://doi.org/10.1128/mBio.00037-15. (In eng).
Malczynski M, Zhu A, Zembower T, Qi C. Diagnostic performance of Ion 16S metagenomics kit and Ion reporter metagenomics workflow for bacterial pathogen detection in culture-negative clinical specimens from sterile sources. Diagn Microbiol Infect Dis. 2021;101(2):115451. DOI:https://doi.org/10.1016/j.diagmicrobio.2021.115451. (In eng).
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200. DOI:https://doi.org/10.1093/bioinformatics/btr381. (In eng).
Edgar RC. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016:081257. DOI:https://doi.org/10.1101/081257.
Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. DOI:https://doi.org/10.7717/peerj.2584. (In eng).
Glöckner FO, Yilmaz P, Quast C, et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol. 2017;261:169–76. DOI:https://doi.org/10.1016/j.jbiotec.2017.06.1198. (In eng).
DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–72. DOI:https://doi.org/10.1128/aem.03006-05. (In eng).
Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12(6):R60. DOI:https://doi.org/10.1186/gb-2011-12-6-r60. (In eng).
Ward T, Larson J, Meulemans J, et al. BugBase predicts organism-level microbiome phenotypes. bioRxiv 2017:133462. DOI: https://doi.org/10.1101/133462.
Douglas G, Maffei V, Zaneveld J, et al. PICRUSt2: An improved and extensible approach for metagenome inference2019.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):11. DOI:https://doi.org/10.1186/1752-0509-8-s4-s11. (In eng). ; Suppl 4 ) .
Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504. DOI:https://doi.org/10.1101/gr.1239303. (In eng).
Wallen ZD. Comparison study of differential abundance testing methods using two large Parkinson disease gut microbiome datasets derived from 16S amplicon sequencing. BMC Bioinformatics. 2021;22(1):265. DOI:https://doi.org/10.1186/s12859-021-04193-6.
Gao L, Xu T, Huang G, Jiang S, Gu Y, Chen F. Oral microbiomes: more and more importance in oral cavity and whole body. Protein Cell. 2018;9(5):488–500. DOI:https://doi.org/10.1007/s13238-018-0548-1. (In eng).
Sotozono M, Kuriki N, Asahi Y, et al. Impact of sleep on the microbiome of oral biofilms. PLoS ONE. 2021;16(12):e0259850. DOI:https://doi.org/10.1371/journal.pone.0259850. (In eng).
Bowerman KL, Rehman SF, Vaughan A, et al. Disease-associated gut microbiome and metabolome changes in patients with chronic obstructive pulmonary disease. Nat Commun. 2020;11(1):5886. DOI:https://doi.org/10.1038/s41467-020-19701-0. (In eng).
Feng XH, Zhang L, Meng HX, Xu L, Chen ZB, Shi D. [Prevalence of putative periodontal microorganisms in Chinese patients with aggressive periodontitis]. Zhonghua Kou Qiang Yi Xue Za Zhi. 2006;41(6):344–7. (In chi).
Guan XY, Liao YT, He L, Meng HX, Li P. [A cross-sectional study of periodontal pathogens in saliva of edentulous patients]. Zhonghua Kou Qiang Yi Xue Za Zhi. 2021;56(12):1230–6. DOI:https://doi.org/10.3760/cma.j.cn112144-20210305-00102. (In chi).
Ma L, Pan YP, Zhang JQ. [Detection of putative periodontal pathogens of periodontitis with type 2 diabetes]. Zhonghua Kou Qiang Yi Xue Za Zhi. 2010;45(6):337–41. (In chi).
Skochko OV, Bobrova NA, Izmaylova OV, Kaĭdashev IP. [Role of several periodontopathogenic microorganisms and tlr4 gene Asp299Gly polymorphism in atherosclerosis pathogenesis]. Zh Mikrobiol Epidemiol Immunobiol 2011(5):83–6. (In rus).
Enigk K, Jentsch H, Rodloff AC, Eschrich K, Stingu CS. Activity of five antimicrobial peptides against periodontal as well as non-periodontal pathogenic strains. J Oral Microbiol. 2020;12(1):1829405. DOI:https://doi.org/10.1080/20002297.2020.1829405. (In eng).
Faveri M, Miquelleto DEC, Bueno-Silva B, et al. Antimicrobial effects of a pulsed electromagnetic field: an in vitro polymicrobial periodontal subgingival biofilm model. Biofouling. 2020;36(7):862–9. DOI:https://doi.org/10.1080/08927014.2020.1825694. (In eng).
Kato I, Vasquez A, Moyerbrailean G, et al. Nutritional Correlates of Human Oral Microbiome. J Am Coll Nutr. 2017;36(2):88–98. DOI:https://doi.org/10.1080/07315724.2016.1185386. (In eng).
Mason MR, Nagaraja HN, Camerlengo T, Joshi V, Kumar PS. Deep sequencing identifies ethnicity-specific bacterial signatures in the oral microbiome. PLoS ONE. 2013;8(10):e77287. DOI:https://doi.org/10.1371/journal.pone.0077287. (In eng).
Fiorillo L. We Do Not Eat Alone: Formation and Maturation of the Oral Microbiota. Biology (Basel) 2020;9(1) (In eng). DOI:https://doi.org/10.3390/biology9010017.
Yousefi L, Leylabadlo HE, Pourlak T, et al. Oral spirochetes: Pathogenic mechanisms in periodontal disease. Microb Pathog. 2020;144:104193. DOI:https://doi.org/10.1016/j.micpath.2020.104193. (In eng).
Goetting-Minesky MP, Godovikova V, Fenno JC. Approaches to Understanding Mechanisms of Dentilisin Protease Complex Expression in Treponema denticola. Front Cell Infect Microbiol. 2021;11:668287. DOI:https://doi.org/10.3389/fcimb.2021.668287. (In eng).
Visser MB, Ellen RP. New insights into the emerging role of oral spirochaetes in periodontal disease. Clin Microbiol Infect. 2011;17(4):502–12. DOI:https://doi.org/10.1111/j.1469-0691.2011.03460.x. (In eng).
Short B, Carson S, Devlin AC, et al. Non-typeable Haemophilus influenzae chronic colonization in chronic obstructive pulmonary disease (COPD). Crit Rev Microbiol. 2021;47(2):192–205. DOI:https://doi.org/10.1080/1040841x.2020.1863330. (In eng).
Dousha L, Sharma R, Lim S, Ngui J, Buckle AM, King PT. Assessing Respiratory Immune Responses to Haemophilus Influenzae. J Vis Exp 2021(172) (In eng). DOI: https://doi.org/10.3791/62572.
Reyes MP, Reyes KC. Gram-negative endocarditis. Curr Infect Dis Rep. 2008;10(4):267–74. DOI:https://doi.org/10.1007/s11908-008-0044-5. (In eng).
Ambrosioni J, Martinez-Garcia C, Llopis J, et al. HACEK infective endocarditis: Epidemiology, clinical features, and outcome: A case-control study. Int J Infect Dis. 2018;76:120–5. DOI:https://doi.org/10.1016/j.ijid.2018.08.013. (In eng).
Bläckberg A, Morenius C, Olaison L, Berge A, Rasmussen M. Infective endocarditis caused by HACEK group bacteria-a registry-based comparative study. Eur J Clin Microbiol Infect Dis. 2021;40(9):1919–24. DOI:https://doi.org/10.1007/s10096-021-04240-3. (In eng).
Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014;30(21):3123–4. DOI:https://doi.org/10.1093/bioinformatics/btu494. (In eng).
Acknowledgements
The authors wish to thank staff members of Linfen Central Hospital for assistance with samples and data collection.
Funding
This work is funded by the Clinical Medical Research Center of Linfen Central Hospital, Key Medical Research Project of Shanxi Province (No. 2021XM19), and Key Research and Development Plan of Linfen Science and Technology (No. 2111).
Author information
Authors and Affiliations
Contributions
GZC takes responsibility for the content of the manuscript, including the data and analysis. GZC and GSM conceived the study, designed the protocol and supervised the project. ZZX, YWY and LGY collected the clinical data and performed the bioinformatics analyses. HYK, SZM and WJ performed the experiments. YWY, MXX, XJB and ZY contributed to the drafting of the manuscript. ZLL, LSQ and WY contributed to sample collection. YWY and HYK contributed to revising the manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Our study was approved by the ethics committee of Linfen Central Hospital (Ethics Approval No. 2022-5-1) and conformed to the Declaration of Helsinki. Informed consent was obtained from all participants.
Consent for publication
Not Applicable.
Competing interests
The authors declare no conflicts of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Zhixia Zhang, Wenyi Yu, and Guangyao Li have contributed equally to this work and share first authorship.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional File 1:
Supplementary Table S1
Additional File 2:
Supplementary Figure
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Z., Yu, W., Li, G. et al. Characteristics of oral microbiome of healthcare workers in different clinical scenarios: a cross-sectional analysis. BMC Oral Health 22, 481 (2022). https://doi.org/10.1186/s12903-022-02501-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12903-022-02501-x