
Abstract
Background
ACTH-independent macronodular hyperplasia (AIMAH) is an uncommon disorder characterized by massive enlargement of both adrenal glands and hypersecretion of cortisol. Concomitant AIMAH and multiple endocrine neoplasia type1 (MEN1) is rare to our knowledge.
Case presentation
Herein, we describe a 32 year old woman with long history of prolactinoma and secondary ammonhrea presented with not-severe manifestation of hypoglycemia due to concomitant presence of insulinoma with AIMAH leading to 12 years delay of MEN1 diagnosis. Laboratory tests showed severe hypoglycemia associated with hyper insulinemia (non-fasting blood sugar = 43 mg/dl, insulin = 80.6 μIU /ml, C-peptide = 9.3 ng/ml) hyperparathyroidism (calcium = 10.3 mg/dl, phosphor = 3.1 mg/dl, PTH = 280 pg/ml) and chemical evidence of an ACTH-independent hypercortisolism (serum cortisol value of 3.5, after 1 mg dexamethasone suppression test serum ACTH value of 17 pg/ml, and high urinary cortisol level). Abdominal CT scan demonstrated two enhancing well-defined masses 27*20 mm and 37*30 mm in the tail and body of the pancreas, respectively, and a 36*15 mm mass in left adrenal gland (seven Hounsfield units). Dynamic pituitary MRI revealed a partial empty sella. The physical examination of the patient was unremarkable. Distal pancreatectomy and a left adrenalectomy were performed. After the surgery, we observed clinical and biochemical remission of hyper insulinemia and gradual decrease in urinary cortisol. The histological features of the removed left adrenal gland were consistent with AIMAH. Histological examination of the pancreatic lesions revealed well differentiated neuroendocrine tumors. Genetic abnormalities in the MEN1, heterozygote for pathogenic variant chr11; 645,773,330-64577333AGAC, c.249-252delGTCT, p. (11e85Serfs Ter33) in exon 2 were found. It was recommended the patient undergoes parathyroidectomy as soon as possible.
Conclusion
Given the history and presentation of our case, we recommend that the clinicians consider the possibility of autonomous cortisol production in MEN1 patients who do not show severe symptoms of hypoglycemia in the presence of insulinoma.
Similar content being viewed by others


Background
ACTH- independent macronodular adrenal hyperplasia (AIMAH) is a rare disorder characterized by enormous bilateral enlargement of adrenal glands with hypercortisolism [1, 2]. AIMAH comprises of approximately 1% of ACTH-independent Cushing syndrome (CS) cases, and the majority of them present with overt CS [3]. Most AIMAH cases are sporadic, and only few such cases with a positive family history have been reported. Although familial cases showed an autosomal dominant inheritance pattern, the genetic mutation has not been identified [4, 5].
Multiple endocrine neoplasia type1 (MEN1), a rare autosomal dominant syndrome, impacts multiple endocrine organs, primarily including parathyroid, anterior pituitary, and pancreas [6]. Adrenal lesions can be detected in 30%—40% of MEN1 patients. Adrenal lesions in MEN1 patients encompass different subtypes from benign adenomas to adrenocortical carcinomas. It has been observed that a significant proportion of adrenal lesions are without any clinical symptoms and functional AIMAH is extremely uncommon. Pancreatic lesions are common in patients with MEN1 and are the main cause of mortality related to the disease [7]. Gastrinoma is the most common pancreatic lesion in MEN-1 patients; however, insulinoma is more common in young patients [8]. We present a case of MEN1 syndrome concomitant with AIMAH that probably masked the severe hypoglycemic manifestations of insulinoma, leading to a 12- year delay of diagnosis.
Case presentation
A 32-year-old woman was referred to the endocrinology clinic for further assessment of recurrent severe hypoglycemia. Menarche occurred at 15 years of age. She reported secondary amenorrhea accompanied by headache and spontaneous galactorrhea from the age of 20. Diagnosis of prolactinoma was confirmed based on high blood prolactin level, and MRI of the pituitary gland and 0.5 mg cabergoline, a dopamine agonist, twice a week was initiated. She became pregnant at age 27 using ovulation induction by Clomiphene, and gave birth to a healthy baby without any complications. Two years after delivery, due to persistent amenorrhea and galactorrhea, a pituitary MRI was performed and showed a large (21*17*14 mm) mass with a leftward deviation of the pituitary stalk. Optic chiasm was intact. During the past year, the patient had several admissions in ED following episodes of severe headache, fatigue, weakness which were reportedly relieved with intravenous dextrose (unfortunately, the data on electrolytes and glucose level were not available). There were no symptoms of hyperandrogenism, weight change, hypertension, and diarrhea.
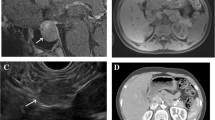
The physical examination of the patient was unremarkable. With suspicion to insulinoma and MEN-1, several blood tests were ordered, which are presented in Table 1. Decreased level of blood sugar and increased level of insulin and C-peptide indicated insulinoma. A raised calcium and PTH level suggested primary hyperparathyroidism Abdominal ultrasound and CT scan were also performed. Ultrasonography revealed two hypoechoic mass-like lesions measuring 27*20 and 10*7 mm near together adjacent to the tail of pancreas, and spiral abdominal CT scan disclosed two enhancing well-defined masses 27*20 mm and 37*30 mm in the tail and body of the pancreases, respectively, calcified areas in larger mass, and a 36*15 mm mass in the left adrenal gland (seven Hounsfield units) (Fig. 1). Results of the blood and urine examinations on second admission are presented in Table 2. By considering MEN1, plasma cortisol after overnight dexamethasone suppression test and urinary free cortisol were requested, which were increased. Also, ACTH level was measured which wasdecreased, indicating primary hypercortisolism. Dynamic pituitary MRI was revealed a partial empty sella (Fig. 2). Bone mineral density (BMD) was performed, which is presented in Table 3. Endoscopic ultrasound (EUS) showed 21*22 mm hypoechoic round lesion with well-defined border in pancreatic body, it was hyper vascular in Doppler and its elastic ratio was 7. Fine needle aspiration (FNA) using EUS from the pancreas masses was performed and an expert pathologist reported neoplastic process with neuroendocrine feature with KI-67 < 2%. Hence, in the patient with the diagnosis of MEN-1 with components of primary hyperparathyroidism, prolactinoma, insulinoma, and CS due to possible adrenal adenoma a spleen-preserving distal pancreatectomy and a left adrenalectomy was performed by a hepatobiliary surgeon. The whole left adrenal gland was enormously enlarged, measuring 100 × 60 × 20 mm. On sectioning, an adrenal tissue with nodular appearance was observed, the maximum of the nodule reached to 60 × 10 mm, which indicated macronodular hyperplasia (H&E,X400, Nikon microscope, Eclipse E100) |(Fig. 3A). In addition, two nodular masses 30 × 30 mm and 20 × 20 mm in the head and tail of pancreas were detected, respectively (H&E, X100, Nikon microscope, Eclipse E100) (Fig. 3B). Tumors were confined to pancreas and all margins were free of tumor. Lymphovascular and perineural invasion were not identified and the tumors stages were T2 NO Mx. After transient hyperglycemia during four days following the surgery that managed with insulin, the level of BS was returned to within reference range. The results of the cortisol and urine free cortisol following the surgery are shown in Table 4. Considering the high level of prolactin at the time of discharge from the hospital (466 ng/ml), low bone mass, the presence of empty sella, and the long duration of amenorrhea, the patient was recommended for 1 mg per week Cabergoline and hormone replacement therapy. Finally, parathyroidectomy was considered for the patients as well.

Without contrast (a) and with contrast (b), axial contrast-enhanced CT of the abdomen shows the hyperenhancing pancreatic body mass (red arrows) as well as indeterminate left adrenal nodules (yellow arrows)

Pituitary MRI revlead partially empty sella

A Macronodular adrenal hyperplasia showing nodules composed of clear and compact cells with variable lipid (H&E,X400, Nikon microscope, Eclipse E100) (B) Normal pancreas( right side) and lesion ( left side) (H&E, X100, Nikon microscope, Eclipse E100)
DNA was extracted from blood cells using salting out method. PCR was used to amplify for 10 coding exons as well as 20 bases of flanking non-coding sequences of MEN1 gene [NM-000244.3]. After cleaning of the PCR products, cycle sequencing was carried out using the ABI Big Dye Terminator v.3.0 kit. Products were resolved by electrophoresis on an ABI 3130 capillary sequencer. Sequencing was performed separately in both the forward and reverse directions. Alignment followed by comparing the individual’s sequences with the reference sequences was performed, respectively. The results indicated that she is heterozygote for pathogenic variant chr11;645,773,330-64577333AGAC, c.249-252delGTCT, p. (11e85Serfs Ter33) in exon 2 of MEN1 gene. This variant has been reported as a pathogenic change in www.ensemble.org and www.ncbi.nlm.nih.gov/clinvar and OSMIC. The above variant has been previously observed in MEN1 patients [9, 10]. Bioinformatics prediction program (Mutation Taster) states that protein features might be affected due to this deletion. Also, according to the American College of Medical Genetics and Genomics (ACMG) standards and guidelines, this variant is classified as pathogenic [11].
There was no remarkable family history except her father had a history of renal stones and lithotripsy; however, his PTH and serum calcium were normal. MEN1 mutations were assessed for her 5-year-old daughter, and the result of the test came back positive (similar mutation to her mother).
Search strategy for literature review
We carried out a data search of PubMed for articles published from January 1990 to February 2021 using the following keywords “MEN-1 and adrenal lesions”, “MEN-1 and AIAMAH”, MEN-1 and autonomous cortisol secretion”, “MEN-1 and cortisol secretion, and MEN-1 and macronodular hyperplasia”. All relevant studies were retrieved, and we also checked their references manually to find any other related papers.
Discussion and conclusion
According to the literature review, corticoadrenal lesions can be detected in 30 to 40% of patients with MEN-1 [12]. A variety of adrenal abnormalities, mainly including non-malignant adenoma and hyperplasia, and less common carcinoma, in patients with MEN-1, can be identified, and the majority of these abnormalities are non-functional [12]. To the best of our knowledge, the concomitant presentation of MEN-1 and macronodular adrenal hyperplasia have been reported in only 11 cases (Table 5) and that four were labeled as AIMAH [12,13,14,15]. Of 4 AIMAH cases, three patients had parathyroid tumors [12, 13, 15], one patient had pituitary tumor (nonfunctional) [12], and two patients had pancreatic tumor (insulinoma) [12, 14].
The most similar case to our patient was described by Yoshida et al. [12]. They described a patient with AIMAH, hyperparathyroidism, pituitary microadenoma, and multifocal insulinoma in addition to diabetes mellitus. All diagnostic tests for Cushing syndrome were normal other than high midnight cortisol concentrations which probably explain insulin resistance and diabetes mellitus. It is noteworthy to mention that our patient, despite repetitive hypoglycemic presentation with weakness, did not report other neuroglycopenic symptoms such as seizure or altered mental status. We assume that autonomous cortisol production by AIMAH can attenuate the hypoglycemic manifestations due to insulin resistance.
In the study of Skogseid et al. [20] and Burgess et al. [16], all MEN-1 patients with adrenal lesions had pancreatic neuroendocrine tumors, similar to our case diagnosed with insulinoma. In addition, it has shown that hyperinsulinemia had some role in adrenocortical tumors development, which probably can be from heterogeneous nature of adrenal glands in proliferation and function [21]. Since the presence of adrenal tumor in sporadic insulinoma is not common, this hypothesis merits further investigations. [17]. It is important to mention that considering the close correlation between neuroendocrine pancreatic tumors and adrenal lesions in MEN 1, abnormality in the genome may be behind this association. Another hypothesis is that hypoglycemia due to hyperinsulinemia, stimulates the secretion of hormones, such as catecholamine and ACTH, which may lead to the proliferation of adrenal cells. However, this theory is unlikely to explain adrenal hyperplasia in patients with MEN-1, because according to Yoshida’s report [12], adrenal gland growth was continuing after the removal of insulinoma.
Mutations in MEN-1 gene, which is located in 11q13 chromosome, are the main cause of MEN-1 in the majority of the patients, even though it has been demonstrated that those mutations are not found in 10% to 30% of the patients. In addition to MEN-1 mutations, in some cases, gross deletions, p27, and p18 defects may contribute to MEN-1 [9]. In the study of Hsiao et al., among 16 patients with AIMAH, MEN-1 mutation (Pro494Leu) was detected in only one patient [13]. Yoshida et al. found no mutations in MEN-1, p27, and p18 genes [22]. Langer et al. pointed out that MEN1 mutations in exon 2 are expected to be observed more than other mutations. They found MEN1 mutation in exon 2 in out of two patients with macronodular hyperplasia [18]. In the present study, the patient was positive for MEN1 mutation in exon 2 as well.
As we mentioned before, AIMAH is characterized by huge bilateral adrenal enlargement; however, up to now, similar to our case, three cases of unilateral AIMAH has been reported [23,24,25]; therefore, the diagnosis of AIMAH cannot be ruled out based on the absence of bilateral lesions at imaging as it has been emphasized that the diagnosis of AIMAH is mostly based on pathological examinations [26]. Thus, in our case, we cannot rule out absence of nodular changes in right adrenal and the right adrenal involvement may be less prominent than left adrenal to be observed at imaging. Also, according to findings of Sheikh-Ahmad et al., the probability of achieving CS remission of people with AIMAH after unilateral adrenalectomy was 94.4% for years [27]; thus, it is necessary to follow our patient for CS recurrence.
The progression of AIMAH is extremely slow, and it takes years to lose the diurnal plasma ACTH rhythm. In AIMAH, plasma ACTH is intended to be decrease and would not be fully suppressed, especially in those with mild cortisol hypersecretion. Similarly, in our patient, ACTH did not fully suppress. The other reasons can be offered ectopic ACTH production by the adrenal glands [28] or in those with GR loss-of-function mutations [29].
A number of literature proposed bilateral laparoscopic adrenalectomy as the optimal treatment for bilateral AIMAH [30, 31]. A study claimed no death attributed to surgery was found in 45 patients with bilateral AIMAH experiencing bilateral adrenalectomy [31]. Unilateral adrenalectomy due to no need for life-tong steroid replacement was suggested by several studies [32, 33]. In a study with a median of 69 months follow-up, the success rate of unilateral adrenalectomy was estimated to be approximately 93% [33]. Likewise, we performed unilateral adrenalectomy (left-sided) plus distal pancreatectomy for our patient, and after the surgery, we observed a constant reduction in UFC.
Although a number of studies have been dedicated to clarify the mechanism of AIMAH, up to now, the precise mechanism is not elucidated. Considering the relevant studies, the pathogenesis of AIMAH can be heterogeneous [1, 34, 35]. While clonal studies reveal that polyclonal pattern can be involved in AIMAH genesis [35], other studies demonstrate that the pathogenesis behind the disease is related to cAMP/protein kinase A pathway [36]. Furthermore, it has been hypothesized that prolonged stimulation of aberrant receptors expressed within the adrenal cortex through several ligands may explain this adrenal abnormality [1, 2].
In the current study, we describe the case of MEN1 with simultaneous presentation of insulinoma and AIMAH that was demonstrated by hormonal and pathological evaluations. Considering the history of our case, we suggest clinicians think of autonomous cortisol production in MEN1 patients who do not show severe symptoms of hypoglycemia in the presence of insulinoma (s).
Availability of data and materials
All data used during the current study are available from the corresponding author on reasonable request.
Abbreviations
- AIMAH:
-
ACTH-independent macronodular adrenal hyperplasia
- MEN1:
-
Multiple Endocrine Neoplasia MEN1
- CS:
-
Cushing Syndrome
- ACTH:
-
Adrenocorticotrophic, Hormone
- LH:
-
Luteinizing Hormone
- FSH:
-
Follicle-stimulating Hormone
- PTH:
-
Parathyroid Hormone
- TSH:
-
Thyroid-stimulating Hormone
- DHEAS:
-
Dehydroepiandrosterone Sulfate
- ED:
-
Emergency Department
- MRI:
-
Magnetic Resonance Imaging
- CT scan:
-
Computed Tomography scan
- EUS:
-
Endoscopic Ultrasound
- FNA:
-
Fine Needle Aspiration
References
Lacroix A, Bourdeau I. Bilateral adrenal Cushing’s syndrome: macronodular adrenal hyperplasia and primary pigmented nodular adrenocortical disease. Endocrinol Metab Clin. 2005;34(2):441–58.
Stratakis CA, Boikos SA. Genetics of adrenal tumors associated with Cushing’s syndrome: a new classification for bilateral adrenocortical hyperplasias. Nat Clin Pract Endocrinol Metab. 2007;3(11):748–57.
Ohashi A, Yamada Y, Sakaguchi K, Inoue T, Kubo M. FUSHIMI H: A natural history of adrenocorticotropin-independent bilateral adrenal macronodular hyperplasia (AIMAH) from preclinical to clinically overt Cushing’s syndrome. Endocr J. 2001;48(6):677–83.
Findlay JC, Sheeler LR, Engeland WC, Aron DC. Familial adrenocorticotropin-independent Cushing’s syndrome with bilateral macronodular adrenal hyperplasia. J Clin Endocrinol Metab. 1993;76(1):189–91.
Lee S, Hwang R, Lee J, Rhee Y, Kim DJ. Chung Ui, Lim SK: Ectopic expression of vasopressin V1b and V2 receptors in the adrenal glands of familial ACTH-independent macronodular adrenal hyperplasia. Clin Endocrinol. 2005;63(6):625–30.
Van Leeuwaarde RS, Van Nesselrooij BP, Hermus AR, Dekkers OM, De Herder WW, Van Der Horst-Schrivers AN, Drent ML, Bisschop PH, Havekes B, Vriens MR. Impact of delay in diagnosis in outcomes in MEN1: results from the Dutch MEN1 study group. J Clin Endocrinol Metab. 2016;101(3):1159–65.
Åkerström G, Hessman O, Hellman P, Skogseid B. Pancreatic tumours as part of the MEN-1 syndrome. Best Pract Res Clin Gastroenterol. 2005;19(5):819–30.
Grant CS. Insulinoma. Best Pract Res Clin Gastroenterol. 2005;19(5):783–98.
Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat. 2008;29(1):22–32.
Thakker RV. Multiple endocrine neoplasia type 1 (MEN1). Best Pract Res Clin Endocrinol Metab. 2010;24(3):355–70.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–23.
Yoshida M, Hiroi M, Imai T, Kikumori T, Himeno T, Nakamura Y, Sasano H, Yamada M, Murakami Y, Nakamura S. A case of ACTH-independent macronodular adrenal hyperplasia associated with multiple endocrine neoplasia type 1. Endocr J. 2011;58(4):269–77.
Hsiao H-P, Kirschner LS, Bourdeau I, Keil MF, Boikos SA, Verma S, Robinson-White AJ, Nesterova M, Lacroix A, Stratakis CA. Clinical and genetic heterogeneity, overlap with other tumor syndromes, and atypical glucocorticoid hormone secretion in adrenocorticotropin-independent macronodular adrenal hyperplasia compared with other adrenocortical tumors. J Clin Endocrinol Metab. 2009;94(8):2930–7.
Lee KT-K, Arnott RD, McLean CA, Serpell JW, Topliss DJ. Corticotropin-independent macronodular adrenal hyperplasia associated with insulinoma. Endocrine Pract. 2011;17(3):e43–7.
Sato M, Soma M, Nakayama T, Kosuge K, Suzuki R, Okada K, Komatsu K, Sugitani M, Matsumoto K. A case of adrenocorticotropin-independent bilateral adrenal macronodular hyperplasia (AIMAH) with primary hyperparathyroidism (PHPT). Endocr J. 2006;53(1):111–7.
Burgess JR, Harle RA, Tucker P, Parameswaran V, Davies P, Greenaway TM, Shepherd JJ. Adrenal lesions in a large kindred with multiple endocrine neoplasia type 1. Arch Surg. 1996;131(7):699–702.
Barzon L, Pasquali C, Grigoletto C, Pedrazzoli S, Boscaro M. FALLO F: Multiple endocrine neoplasia type 1 and adrenal lesions. J Urol. 2001;166(1):24–7.
Langer P, Cupisti K, Bartsch DK, Nies C, Goretzki PE, Rothmund M, Röher HD. Adrenal involvement in multiple endocrine neoplasia type 1. World J Surg. 2002;26(8):891–6.
Waldmann J, Bartsch D, Kann P, Fendrich V, Rothmund M, Langer P. Adrenal involvement in multiple endocrine neoplasia type 1: results of 7 years prospective screening. Langenbecks Arch Surg. 2007;392(4):437–43.
Skogseid B, Larsson C, Lindgren P, Kvanta E, Rastad J, Theodorsson E, Wide L, Wilander E, Oberg K. Clinical and genetic features of adrenocortical lesions in multiple endocrine neoplasia type 1. J Clin Endocrinol Metab. 1992;75(1):76–81.
Tsatsoulis A. The role of insulin resistance/hyperinsulinism on the rising trend of thyroid and adrenal nodular disease in the current environment. J Clin Med. 2018;7(3):37.
Conemans EB, Raicu-Ionita G, Pieterman C, Dreijerink K, Dekkers O, Hermus A, De Herder W, Drent M, Van Der Horst-Schrivers A, Havekes B. Expression of p27 Kip1 and p18 Ink4c in human multiple endocrine neoplasia type 1-related pancreatic neuroendocrine tumors. J Endocrinol Invest. 2018;41(6):655–61.
Bertagna X, Groussin L, Luton J-P, Bertherat J. Aberrant receptor-mediated Cushing’s syndrome. Hormone Res Paediatrics. 2003;59(Suppl. 1):99–103.
Groussin L, Perlemoine K, Contesse V, Lefebvre H, Tabarin A, Thieblot P, Schlienger JL, Luton JP, Bertagna X. Bertherat Jrm: The ectopic expression of the gastric inhibitory polypeptide receptor is frequent in adrenocorticotropin-independent bilateral macronodular adrenal hyperplasia, but rare in unilateral tumors. J Clin Endocrinol Metab. 2002;87(5):1980–5.
Rosenberg D, Groussin L, Bertagna X, Bertherat J. cAMP pathway alterations from the cell surface to the nucleus in adrenocortical tumors. Endocr Res. 2002;28(4):765–75.
Li J, Yang CH. Diagnosis and treatment of adrenocorticotrophic hormone-independent macronodular adrenocortical hyperplasia: a report of 23 cases in a single center. Exp Ther Med. 2015;9(2):507–12.
Sheikh-Ahmad M, Dickstein G, Matter I, Shechner C, Bejar J, Reut M, Sroka G, Laniado M, Saiegh L. Unilateral adrenalectomy for primary bilateral macronodular adrenal hyperplasia: analysis of 71 cases. Exp Clin Endocrinol Diabetes. 2020;128(12):827–34.
Louiset E, Duparc C, Young J, Renouf S, Tetsi Nomigni M, Boutelet I, Libé R, Bram Z, Groussin L, Caron P. Intraadrenal corticotropin in bilateral macronodular adrenal hyperplasia. N Engl J Med. 2013;369(22):2115–25.
Vitellius G, Trabado S, Hoeffel C, Bouligand J, Bennet A, Castinetti F, Decoudier B, Guiochon-Mantel A, Lombes M, Delemer B. Significant prevalence of NR3C1 mutations in incidentally discovered bilateral adrenal hyperplasia: results of the French MUTA-GR Study. Eur J Endocrinol. 2018;178(4):411–23.
Aggarwal S, Yadav K, Sharma AP, Sethi V. Laparoscopic bilateral transperitoneal adrenalectomy for Cushing syndrome: surgical challenges and lessons learnt. Surgical Laparoscopy Endoscopy & Percutaneous Techniques. 2013;23(3):324–8.
Ritzel K, Beuschlein F, Mickisch A, Osswald A, Schneider HJ, Schopohl J, Reincke M. Outcome of bilateral adrenalectomy in Cushing’s syndrome: a systematic review. J Clin Endocrinol Metab. 2013;98(10):3939–48.
Kobayashi T, Miwa T, Kan K, Takeda M, Sakai H, Kanazawa A, Tanaka A, Namiki K, Nagao T, Odawara M. Usefulness and limitations of unilateral adrenalectomy for ACTH-independent macronodular adrenal hyperplasia in a patient with poor glycemic control. Intern Med. 2012;51(13):1709–13.
Xu Y, Rui W, Qi Y, Zhang C, Zhao J, Wang X, Wu Y, Zhu Q, Shen Z, Ning G. The role of unilateral adrenalectomy in corticotropin-independent bilateral adrenocortical hyperplasias. World J Surg. 2013;37(7):1626–32.
Bourdeau I, Antonini SR, Lacroix A, Kirschner LS, Matyakhina L, Lorang D, Libutti SK, Stratakis CA. Gene array analysis of macronodular adrenal hyperplasia confirms clinical heterogeneity and identifies several candidate genes as molecular mediators. Oncogene. 2004;23(8):1575–85.
Gicquel C, Leblond-Francillard M, Bertagna X, Louvel A, Chapuis Y, Luton JP, Girard F, Le Bouc Y. Clonal analysis of human adrenocortical carcinomas and secreting adenomas. Clin Endocrinol (Oxf). 1994;40(4):465–77.
Bourdeau I, Parisien-La Salle S, Lacroix A. Adrenocortical hyperplasia: A multifaceted disease. Best Pract Res Clin Endocrinol Metab. 2020;34(3):101386.
Acknowledgements
We express our appreciation to the research team members and to TLGS participants for their contribution to the study.
Funding
None.
Author information
Authors and Affiliations
Contributions
FH, SSTZ, and VJ wrote the manuscript, AR: performed the surgery, SAHM reported the pathology of the patient; ZM performed and interpreted the sonography and MRI, FH and SK reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
An approval from an ethics committee was not needed for this case report since it involved one patient.
Consent for publication
Written informed consent was obtained from the patient for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Series Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chavoshi, V., Tamehri Zadeh, S.S., Khalili, S. et al. Long delay in diagnosis of a case with MEN1 due to concomitant presence of AIMAH with insulinoma: a case report and literature review. BMC Endocr Disord 22, 108 (2022). https://doi.org/10.1186/s12902-022-01022-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12902-022-01022-6