
Abstract
Background
Manifestations of hypokalaemia in ectopic adrenocorticotropic hormonesyndrome(EAS) vary from mild muscle weakness to life-threatening arrhythmia. Herein, we present a rare case of EAS with concomitant rhabdomyolysis(RM) as a result of intractable hypokalaemia.
Case presentation
A 64-year-old man was admitted for limb weakness and facial hyperpigmentation for 2 weeks. Lab tests revealed intractable hypokalaemia (lowest at 1.8 mmol/L) and metabolic alkalosis. The diagnosis of RM was based on a creatine kinase(CK)level of 5 times the upper limit. The elevated CK and myohemoglobin (Mb) levels returned to within the normal range after the alleviation of hypokalaemia. The patient was diagnosed with ACTH-dependent Cushing’s syndrome (CS) based on unsuppressed serum cortisol after a low-dose dexamethasone suppression test(LDDST) and remarkably elevated ACTH levels. The diagnosis of EAS was made based on the results of a high-dose dexamethasone suppression test(HDDST) and bilateral inferior petrosal sinus sampling(BIPSS). Multiple lymph nodes in the left supraclavicular fossa, right root of neck, mediastinum and bilateral hili of the lung were found with abnormal uptake of 68Ga-DOTA-NOC. Mediastinoscopic lymph node biopsy was performed. The pathological diagnosis was small-cell and large-cell neuroendocrine carcinoma with positive ACTH staining. The patient was prescribed mifepristone and received one cycle of chemotherapy. The patient could not tolerate subsequent chemotherapy and died of dyscrasia.
Conclusions
RM is a rare complication of EAS with insidious onset and atypical clinical manifestations. Serum potassium levels should be vigilantly monitored to avoid RM in EAS.
Similar content being viewed by others

Background
Cushing’s syndrome (CS) is a set of clinical syndromes caused by excessive production of cortisol. Ectopic adrenocorticotropic hormone syndrome (EAS) accounts for approximately 12-17 % of CS cases in which ACTH is produced by tumours [1, 2]. Diagnosis and treatment are more challenging for EAS than for other types of CS. The classical features of CS, such as moon face, plethora and central obesity, are absent in patients with aggressive tumours of short duration and cachexia. The localization of the tumours is sometimes time consuming or ultimately futile, even when functional imaging is available. Additionally, extreme hypercortisolism results in a high risk of life-threatening opportunistic infections and overwhelming biochemical disorders, among which hypokalaemia is the most prominent. The manifestations of hypokalaemia vary greatly from mild muscle weakness to life-threatening arrhythmia. Herein, we present a rare case of EAS with concomitant rhabdomyolysis(RM) resulting from intractable hypokalaemia.
Case presentation
A 64-year-old man was admitted to a local hospital for limb weakness and facial hyperpigmentation for 2 weeks. He also reported oedema of the lower limbs for 1 week. He had a history of smoking for 40 years(30 cigarettes per day). Lab tests revealed severe hypokalaemia (1.8 mmol/L, normal range 3.5–5.5 mmol/L) and metabolic alkalosis. Potassium supplementation (KCl, 9 g/day) failed to normalize the serum potassium (peaked at 2.3 mmol/L). As the level of serum creatine kinase (CK) was remarkably elevated(1268.4 u/L, normal range 50–200 u/L), the patient was transferred to our hospital. Upon admission, mild moon face and facial hyperpigmentation was noticed when compared with photos taken 2 years before. Oedema of the lower extremities and decreased muscle strength were also found during physical examination. Hyperpigmentation at interphalangeal joints and ecchymosis gradually developed during hospitalization.
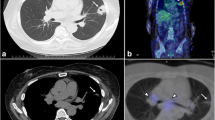
The results of the laboratory test on admission and the function of the endocrine glands are summarized in Tables 1 and 2. Urinary potassium excretion was inappropriately increased (Table 1). The serum cortisol level was increased, and diurnal rhythm was lost. The level of serum cortisol at 8 am was > 50 µg/dL after the low-dose dexamethasone suppression test(LDDST), and the patient was diagnosed with CS. ACTH dependency was confirmed based on remarkably elevated ACTH levels (Table 2). Pituitary MR was normal (Fig. 1a and b). The serum cortisol level at 8 am before and after the high-dose dexamethasone suppression test (HDDST) was 212 µg/dL and 208 µg/dL (calculated with the measured value and dilution rate), respectively (Table 2). Although the above results strongly suggested EAS, bilateral inferior petrosal sinus sampling (BIPSS) was routinely arranged for verification, mainly for two reasons. First, false positive and false negative results occur in both HDDST and regular imaging(pituitary MR and chest CT). For example, the HDDST results for EAS and CD sometimes overlap. Some patients with EAS have coexisting pituitary adenomas that are not the source of ACTH. Second, in very few patients, CD is caused by hyperplasia instead of adenoma, resulting in a negative finding on pituitary MR. Then, BIPSS was successfully performed (PRLleft/PRLperipheral and PRLright/PRLperipheral were 4.3 ± 0.5 and 2.5 ± 0.3, respectively). The left, right and peripheral ACTH levels were 789.9 ± 5.2 pg/mL, 773.6 ± 18.2 pg/mL and 726.5 ± 26.7 pg/mL (three samples for each), respectively, and the left, right and peripheral ACTH/PRL levels were 12.4 ± 1.4, 21.4 ± 2.4 and 49.1 ± 2.2, respectively. The diagnosis of EAS was made based on the HDDST and BIPSS results. Chest and abdominal CT as well as neck ultrasound were further arranged. Abdominal CT showed diffuse enlargement of the bilateral adrenal glands (Fig. 1c and d), and there were no abnormalities in the pancreas. Chest CT indicated two nodules in the superior and middle lobes of the right lung (Fig. 1e f) and prominent mediastinal lymphadenopathy (Fig. 1g h). Elevated tumour biomarkers, including carcinoembryonic antigen (CEA), cytokeratin 21 − 1 fragment CYFRA21-1 and neuron-specific enolase(NSE), were detected (Table 1). Thyroid ultrasound showed no abnormality. 68Ga-PET/CT suggested high-density opacity in the superior lobe of the right lung with a slight abnormally increased uptake of 68Ga-DOTA-NOC, fitting the characteristics of chronic infection (Fig. 1i). The high-density nodule in the middle lobe of the right lung was found without abnormal uptake of 68Ga-DOTA-NOC (Fig. 1j). Multiple lymph nodes in the left supraclavicular fossa, right root of the neck, mediastinum and bilateral hili of the lung were found partially fused into a mass with abnormal uptake of 68Ga-DOTA-NOC (Fig. 1k and l). Considering that the lymph nodes in the supraclavicular fossa and root of the neck might be distant metastases which is differ from the primary lesion, lymph nodes in the mediastinum and bilateral hili were preferable for biopsy. Therefore, mediastinoscopic lymph node biopsy was performed. The lymph nodes were pale and friable (Fig. 2a). The pathological diagnosis was small-cell and large-cell neuroendocrine carcinoma (LCNEC)according to the 2018 World Health Organization expert consensus proposal on the classification framework for neuroendocrine neoplasms [3] (Fig. 2b and c). Mitosis was approximately 80/10 HPF (Fig. 2d), and the Ki-67 index was 80 % (Fig. 2e). ACTH staining was positive (Fig. 2f).

Imaging examinations. Pituitary MRI revealed no abnormalities (a and b). Abdominal CT showed diffuse enlargement of the bilateral adrenal glands (c and d). Chest CT indicated two nodules in the superior (e) and middle lobes of the right lung (f) and mediastinal lymphadenopathy (g and h). 68Ga PET/CT demonstrated high-density opacity in the superior lobe of the right lung with a slight abnormally increased uptake of 68Ga-DOTA-NOC (i). High-density nodules in the middle lobe of the right lung did not show abnormal uptake of 68Ga-DOTA-NOC (j, arrow). Multiple lymph nodes in the mediastinum and bilateral hili of the lung partially fused into a mass with abnormally increased uptake of 68Ga-DOTA-NOC (k and l)

Mediastinoscopy revealed enlarged lymph nodes that were pale and friable (a). Large tumour cells with moderate to abundant cytoplasm and prominent nucleoli (b). Densely packed small tumour cells with scant cytoplasm, finely granular nuclear chromatin and absent nucleoli (c). Mitosis (d). Ki-67 index was 80 % (e). Immunohistochemical staining for ACTH (f). b, c and d, HE staining, 400×; e and f, 200×
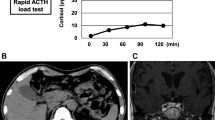
The fasting plasma glucose (FPG) level was 7.4 mmol/L, and the glycosylated haemoglobin(HbA1c) level was 5.7 % on admission. During hospitalization, the FPG level gradually increased. Diagnosis of diabetes was made, and insulin therapy was initiated. Bone mineral density indicated low bone mass. Blood pressure, lipid profile, and coagulation function were generally normal.No thrombus was found by colour Doppler ultrasound in the arteries or veins of the lower extremities. On admission, remarkably elevated levels of serum CK and myohemoglobin (Mb)were noticed with slightly increased creatine kinase isoenzyme(CKMB) (Table 1). ECG was generally normal. Based on the above results and the even higher CK level (higher than 5 times the upper limit) at the local hospital, the diagnosis of RM was made, and fluid infusion was applied. The serum potassium level was 2.49 mmol/L on admission, and potassium supplementation (KCl, 15 g/d) was initiated. After the assessment of renin, angiotensin and aldosterone, antisterone was prescribed at a daily dose of 120–160 mg. The CK and Mb levels decreased gradually as the serum potassium level increased. On day 10, a saline and glucose solution was applied after the enhanced pituitary MR and chest CT to prevent acute kidney injury, and transient decline in the serum potassium level and re-elevation of the CK and Mb levels occurred (Fig. 3a, b and c). On admission, an increased troponin T level, a decreased serum calcium level and an increased parathyroid hormone(PTH)level were also observed and were gradually relieved with the improvement in RM.

Serum potassium level and corresponding treatment (a). Fluctuation of serum CK and Mb levels (b and c). The space between dashed lines (a) or between the dashed line and the x-axis (b and c) is the normal range
After discussions among specialists from multiple disciplines (pathology, respiratory medicine, oncology, anaesthesiology, urology, thoracic surgery and endocrinology department), the urologists and anaesthetists considered it very risky for the patient to undergo bilateral adrenalectomybased on hypokalaemia, infection and general status. Additionally, aggravation of the condition was more related to malignancy than hypercortisolaemia, especially after the application of mifepristone. As the priority was determined to be to treat neuroendocrine carcinoma, which was unresectable, the patient was transferred to the oncology department to receive chemotherapy. In addition, mifepristone was applied at an initiating dosage of 75 mg per day and a maintaining dosage of 150 mg per day. The patient received one cycle of chemotherapy with the EP regimen (etoposide 100 mg iv drip days 1–3 and cisplatin days 1–3) but could not tolerate subsequent chemotherapy. He died of dyscrasia within 1 month.
Discussion and conclusion
We reported a representative case of EAS caused by a very aggressive malignancy, with atypical manifestation of CS and no opportunity for radical resection at diagnosis. As a rare cause of CS, EAS has always been a clinical challenge. First, EAS often has an insidious onset. Typical signs of CS, such as moon face and central obesity, are often absent from the influence of cachexia, leading to delayed diagnosis. Second, some tumours are highly malignant and progress rapidly, and diagnosis and treatment are urgently needed. The diagnosis from ACTH-dependent CS to the specific causes was roughly divided into two steps. The first step is to distinguish whether the lesion is located in the centre (pituitary or hypothalamus) or the periphery. The amplitude of ACTH and the HDDST, pituitary MR, chest and abdomen CT results can be suggestive. However, as the incidence of incidental pituitary adenomas is as high as 10 %, identification of a pituitary adenoma does not always guarantee the identification of the responsible lesion. Moreover, the ACTH levels and HDDST results of some peripheral carcinoid and pituitary adenomas overlap. Some tumour biomarkers are not specific and increase in chronic inflammation. With the combination of HDDST and BIPSS, 95 % of the cases can be distinguished [2, 4, 5]. However, due to the technical skills required to perform BIPSS and vascular variation, this procedure sometimes fails, and the results can be ambiguous in certain cases. The ultimate challenge is the localization of the tumour. EAS is most frequently caused by lung or mediastinal carcinoid and small-cell lung cancer(SCLC). The less common causes include insulinoma, medullary thyroid cancer, gastrointestinal neuroendocrine tumour and pheochromocytoma [2]. Therefore, chest CT should be applied first, and other imaging modalities, including abdominal CT and thyroid ultrasound, could be arranged. One difficulty is that the high incidence/detection rate of pulmonary nodules results in more than one suspected lesion in some cases. As in this patient, two nodules were found in the right lung, while enlarged lymph nodes were found in the mediastinum and both hilar regions. PET/CT can be further applied if CT fails. Specifically, 68Ga PET/CT is effective in locating pulmonary neuroendocrine tumours, while 18-F FDP is suitable for malignancies known or speculated to be more aggressive. For 68Ga and 18-F FDP PET/CT-negative cases, somatostatin imaging can be adopted, but persistent hypercorticosteraemia may suppress the expression of the somatostatin subtype 2 receptor and cause false-negative results in some cases [6]. Dopamine receptor PET/CT is used mainly to locate tumours with co-secretion of catecholamine and ACTH. For cases with negative functional imaging, segmental body vein blood sampling can theoretically be used for localization but is of limited value for adjacent lesions sharing the same drainage vein. Fibrobronchoscopic and percutaneous transthoracic biopsy can be applied to verify the suspected lesions in the lung, and endobronchial ultrasound and mediastinoscopy biopsies can be applied for those in the mediastinum. For our patient, either endobronchial ultrasound or mediastinoscopy biopsies were feasible, and mediastinoscopy biopsy was ultimately opted to obtain sufficient tissue and to facilitate haemostasis under direct vision in case of intraoperative bleeding.
The ultimate cure for patients with EAS is radical resection of the malignancy, which was not feasible for our patient who had multiple metastases. LCNEC and small-cell carcinoma are both high-grade neuroendocrine carcinomas. Early surgical resection is the first-line therapy for LCNEC, and the 5-year and 10-year survival rates are approximately 30 and 10 %, respectively. Most patients with small-cell carcinoma require chemotherapy or a combination of chemotherapy and radiotherapy, and the 5-year and 10-year survival rates are approximately 10 % and less than 5 %, respectively [7]. For EAS patients with unresectable lesions, medicines have been reported to be effective in normalizing cortisol levels in 83.3 %(64.9–96.8) of patients [8]. Since neither mitotane nor cabergoline are marketed in China, the glucocorticoid receptor antagonist mifepristone is also used for palliative therapy.
This patient had RM caused by extreme hypokalaemia, a rare complication of EAS. There are diverse causes of rhabdomyolysis. Patients with CS are susceptible to infection due to immunodeficiency caused by hypercortisolaemia, which is a common aetiology for RM. Disturbances of electrolytes such as hyponatraemia, hypokalaemia and hypophosphataemia should also be considered in the aetiological diagnosis of RM [9]. For the former, some tumours causing EAS may secrete peptides causing SIADH. Pulmonary infection is also common in patients with EAS and may also result in SIADH [10, 11]. Our patient did not exhibit the manifestations of SIADH. Both the serum sodium level and the osmotic pressure were generally normal during hospitalization, ruling out SIADH. This patient had mild hypophosphataemia. However, based on the severity of hypophosphatemia and the reversing trend of the levels of CK and K+, we considered hypokalaemia instead of hypophosphatemia to result in RM.In patients with CS, extra cortisol binds to mineralocorticoid receptors and leads to hypokalaemia. Some medications, such as diuretics and mifepristone, contribute to aggravation. Hypokalaemia in patients with EAS or adrenocortical carcinoma(ACC) is often intractable due to the extremely high cortisol level. K+ regulates blood flow in muscle tissue by dilating vessels. As skeletal muscles consume large amounts of oxygen, hypokalaemia impairs blood supply and causes ischaemic damage. Hypokalaemia also increases the permeability of the myolemma by disturbing transmembrane ion transport [12]. The severity of muscle injury is closely related to plasma potassium levels. Myasthenia and myalgia may be reported when the K+level is below 3.0 mmol/L, elevation of myoenzymes may occur when the K+level is below 2.5 mmol/L, and RM might occur when the K+level is below 2.0 mmol/L [9, 12].
An interesting issue is that RM is reported far more frequently in primary aldosteronism (PA), although PA and CS are both related to severe hypokalaemia. One explanation is the very low incidence of EAS and ACC. Another possible mechanism is that hypokalaemia-induced RM is caused by the imbalance of oxygen supply and consumption. Patients with EAS and ACC usually have significant protein catabolism and muscle loss due to extreme hypercorticosteraemia. Many of them also have restricted physical activities due to pathological fracture or cachexia. As oxygen consumption is determined by muscle content and activity status (resting or exercising), patients with CS have a lower oxygen requirement. In contrast, muscle content and physical activity are generally normal in patients with PA, making them more vulnerable to impaired blood and oxygen supply caused by hypokalaemia.
Clinical manifestations of RM vary greatly. The typical triad of myasthenia, myalgia and coloured urine occurs in less than 10 % of patients, while more than 50 % of patients have no symptoms or signs [12,13,14,15]. Endocrine disease-related RM is even more insidious. Muscle enzymes should be measured, and RM should be considered in patients with CS or PA when the serum potassium level is below 2.0 mmol/l. From a monitoring aspect, since Mb can be eliminated within 24 h from serum, its re-elevation suggests new onset of muscle damage [16]. The treatment of RM should be focused on fluid replacement and the consequent adjustment of potassium supplementation. For CS patients with RM, hypokalaemia and metabolic alkalosis to some extent alleviate the hyperkalaemia and hyperlactacidaemia caused by RM, and complications, including acute renal injury and arrhythmia, are rare. It should also be noted that with the destruction of muscle cells, a large amount of phosphate is released that binds to calcium ions and accumulates in the damaged muscle, resulting in hypocalcaemia [17], as was observed in this patient. PTH elevation is secondary to hypocalcaemia, which does not need to be treated. Calcium supplementation is also unnecessary and might cause ectopic calcification in muscle tissue [18].
In summary, RM is a rare complication of EAS with atypical clinical manifestations. Serum potassium levels should be vigilantly monitored to avoid RM in EAS.
Availability of data and materials
All relevant data supporting the conclusions of this article are included within the article.
Abbreviations
- CS:
-
Cushing’s syndrome
- EAS:
-
Ectopic adrenocorticotropic hormonesyndrome
- ACTH:
-
Adrenocorticotropic hormone
- RM:
-
Rhabdomyolysis
- CK:
-
Creatine kinase
- LDDST:
-
Low-dose dexamethasone suppression test
- MR:
-
Magnetic resonance
- HDDST:
-
High-dose dexamethasone suppression test
- BIPSS:
-
Bilateral inferior petrosal sinus sampling
- PRL:
-
Prolactin
- CT:
-
Computed tomography
- CEA:
-
Carcinoembryonic antigen
- CYFRA21-1:
-
Cytokeratin 21 − 1 fragment
- NSE:
-
Neuron specific enolase
- LCNEC:
-
Large-cell neuroendocrine carcinoma
- WHO:
-
World Health Organization
- FPG:
-
Fasting plasma glucose
- HbA1c:
-
Glycosylated haemoglobin
- Mb:
-
Myohemoglobin
- CKMB:
-
Creatine kinase isoenzyme
- ECG:
-
Electrocardiogram
- PTH:
-
Parathyroid hormone
- SIADH:
-
Syndrome of inappropriate antidiuretic hormone
- ACC:
-
Adrenocortical carcinoma
- PA:
-
Primary aldosteronism
References
Ferone D, Albertelli M. Ectopic Cushing and other paraneoplastic syndromes in thoracic neuroendocrine tumors. Torac Surg Clin. 2014;24(3):277–83.
Isidori AM, Lenzi A. Ectopic ACTH syndrome. Arquivos brasileiros de endocrinologia emetabologia. 2007;51(8):1217-25.
Rindi G, Klimstra DS, Abedi-Ardekani B, Asa SL, Bosman FT, Brambilla E, et al. A commonclassification framework for neuroendocrine neoplasms: an International Agencyfor Research on Cancer (IARC) and World Health Organization (WHO) expertconsensus proposal. Mod Pathol. 2018;31(12):1770–86.
Isidori AM, Kaltsas GA, Pozza C, Frajese V, Newell-Price J, Reznek RH, et al. The ectopic adrenocorticotropin syndrome: clinical features, diagnosis, management, and long-term follow-up. J Clin Endocrinol Metab. 2006;91(2):371–7.
Isidori AM, Kaltsas GA, Mohammed S, Morris DG, Jenkins P, Chew SL, et al. Discriminatory value of the low-dose dexamethasone suppression test in establishing the diagnosis and differential diagnosis of Cushing’s syndrome. J Clin Endocrinol Metab. 2003;88(11):5299–306.
Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet. 2015;386(9996):913–27.
Huang Q, Muzitansky A, Mark EJ. Pulmonary neuroendocrine carcinomas. A review of 234 cases and a statistical analysis of 50 cases treated at one institution using a simple clinicopathologic classification. Arch Pathol Lab Med. 2002;126(5):545–53.
Broersen LHA, Jha M, Biermasz NR, Pereira AM, Dekkers OM. Effectiveness of medical treatment for Cushing’s syndrome: a systematic review and meta-analysis. Pituitary. 2018;21(6):631–41.
Allison RC, Bedsole DL. The other medical causes of rhabdomyolysis. Am J MedSci. 2003;326(2):79–88.
Rees JH. Paraneoplastic syndromes: when to suspect, how to confirm, and howto manage. J Neurol Neurosurg Psychiatry. 2004;75(Suppl 2):ii43-50.
Tsoli M, Dimitriadis GK, Androulakis II, Kaltsas G, Grossman A. Paraneoplastic Syndromes Related to Neuroendocrine Tumors. 2020 Sep 26. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, Grossman A,Hershman JM, Hofland J, Kaltsas G, Koch C, Kopp P, Korbonits M, McLachlan R, Morley JE, New M, Purnell J, Singer F, Stratakis CA, Trence DL, Wilson DP, editors. Endotext. South Dartmouth: MDText.com, Inc.; 2000–.
Knochel JP. Neuromuscular manifestations of electrolyte disorders. Am J Med. 1982;72(3):521–35.
Zutt R, van der Kooi AJ, Linthorst GE, Wanders RJA, de Visser M. Rhabdomyolysis: Review of the literature. Neuromuscular Disorders Nmd. 2014;24(8):651-9.
Bosch X, Poch E, Grau JM. Rhabdomyolysis and acute kidney injury. N Engl J Med. 2009;361(1):62–72.
Huerta-Alardín AL, Varon J, Marik PE. Bench-to-bedside review: Rhabdomyolysis -- an overview for clinicians. Crit Care (London England). 2005;9(2):158–69.
Giannoglou GD, Chatzizisis YS, Misirli G. The syndrome of rhabdomyolysis: Pathophysiology and diagnosis. European journal of internal medicine. 2007;18(2):90–100.
SG H. Pathogenesis and treatment of renal dysfunction in rhabdomyolysis. Intensive Care Med. 2001;27(5):803–11.
Parekh R, Care DA, Tainter CR. Rhabdomyolysis: advances in diagnosis and treatment. Emerg Med Pract. 2012;14(3):1–15 quiz 15.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key R&D Program of China(2018YFC1311500), National Nature Science Foundation of China (Grant No. 81802829), and Natural Science Basic Research Plan in Shaanxi Province (Grant No. 2020JQ-501 and 2019JQ-122). No funding body participated in the design of the study; the collection, analysis, and interpretation of the data;or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
WQ, SS and HG collected patient data, obtained consent for publication and wrote the first draft of the article. TC, ZW, JF and JZ participated in the treatment, provided consultation and critically reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The patient gave written informed consent to publish on admission when the diagnosis of Cushing’s syndrome complicated with rhabdomyolysis was made.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Qiang, W., Song, S., Chen, T. et al. A rare case of ectopic ACTH syndrome with rhabdomyolysis. BMC Endocr Disord 21, 98 (2021). https://doi.org/10.1186/s12902-021-00755-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12902-021-00755-0