
Abstract
Background
The prevalence of obesity and its comorbidities, including type 2 diabetes mellitus (T2DM), is dramatically increasing throughout the world; however, the underlying aetiology is incompletely understood. Genome-wide association studies (GWAS) have identified hundreds of genec susceptibility loci for obesity and T2DM, although the causal genes and mechanisms are largely unknown. SPRY2 is a candidate gene identified in GWAS of body fat percentage and T2DM, and has recently been linked to insulin production in pancreatic β-cells. In the present study, we aimed to further understand SPRY2 via functional characterisation in HepG2 cells, an in vitro model of human hepatocytes widely used to investigate T2DM and insulin resistance.
Methods
CRISPR-Cas9 genome editing was used to target SPRY2 in HepG2 cells, and the functional consequences of SPRY2 knockout (KO) and overexpression subsequently assessed using glucose uptake and lipid droplet assays, measurement of protein kinase phosphorylation and RNA sequencing.
Results
The major functional consequence of SPRY2 KO was a significant increase in glucose uptake, along with elevated lipid droplet accumulation. These changes were attenuated, but not reversed, in cells overexpressing SPRY2. Phosphorylation of protein kinases across key signalling pathways (including Akt and mitogen activated protein kinases) was not altered after SPRY2 KO. Transcriptome profiling in SPRY2 KO and mock (control) cells revealed a number of differentially expressed genes related to cholesterol biosynthesis, cell cycle regulation and cellular signalling pathways. Phospholipase A2 group IIA (PLA2G2A) mRNA level was subsequently validated as significantly upregulated following SPRY2 KO, highlighting this as a potential mediator downstream of SPRY2.
Conclusion
These findings suggest a role for SPRY2 in glucose and lipid metabolism in hepatocytes and contribute to clarifying the function of this gene in the context of metabolic diseases.
Similar content being viewed by others


Background
The worldwide prevalence of obesity has markedly increased since 1980, with more than 609 million individuals classified as obese in 2015 [1]. Obesity (defined as a body mass index (BMI) of 30 kg/m2 or higher) is a risk factor for the development of serious chronic diseases, including type 2 diabetes mellitus (T2DM), cardiovascular disease (CVD), non-alcoholic fatty liver disease (NAFLD) and chronic kidney disease [2, 3]. CVD (and indirectly, T2DM) is the leading cause of death and disability due to high BMI [4]. The pathogenesis of obesity is not completely understood. A substantial number of behavioural and lifestyle factors influence susceptibility to obesity; however, the risk conferred by genetic and epigenetic traits is also significant with a heritability of 40–70% [5]. Genome-wide association studies (GWAS) have identified a large number of loci robustly associated with obesity-related traits, such as BMI and body fat percentage (BF%), although the causal genes in these loci remain to be established and characterised.
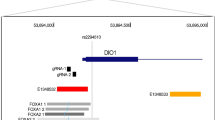
One such locus identified in GWAS meta-analyses of BF% [6, 7] is near the Sprouty RTK signalling antagonist 2 (SPRY2) gene with no other genes within a 1 Mb window around the GWAS signal (Additional file 1: Figure S1). The lead variant (rs534870), 54 kb downstream of SPRY2, exhibited a modest association with BMI, body weight and risk of obesity; importantly, its major allele was associated with a 0.14% decrease in BF% in individuals of European descent [6]. In a larger follow-up study, the lead variant (rs693839, in high linkage equilibrium with rs534870, R2 > 0.95) near SPRY2 was similarly found to have a greater effect on BF% than BMI, suggesting a primary association with adiposity and body fat distribution rather than overall body weight. Additional experiments in Drosophila supported SPRY2 as the likely causal gene [7]. Furthermore, several studies have implicated SPRY2 as a potential candidate gene for T2DM. The rs1359790 variant [8], situated 193 kb upstream to SPRY2, was significantly associated with susceptibility to T2DM in Chinese [9] and Japanese [10] individuals. Sprouty proteins are negative regulators of receptor tyrosine kinase (RTK) signalling pathways [11], which mediate a wide variety of key cellular processes, including critical roles in proliferation, communication and differentiation, as well as influences on motility, metabolism and survival [12]. SPRY2 specifically modulates the Ras/mitogen activated protein (MAP) kinase pathway [13, 14] and may function as a tumour suppressor gene, since its expression has been found to be repressed in a variety of cancers (reviewed in [15]). Other examples of RTK families include vascular endothelial growth factors (VEGF), insulin-like growth factors (IGF), fibroblast growth-factors (FGF) and platelet-derived growth factors (PDGF).
In a recent study utilising whole-genome RNAi [16], SPRY2 was identified as a novel regulator of insulin transcription, and deletion of SPRY2 in adult mouse β-cells led to mild hyperglycaemia and hypoinsulinaemia. However, based on the GWAS findings, there is reason to believe that SPRY2 may also be involved in peripheral insulin resistance, metabolic syndrome or hepatosteatosis, rather than just insulin secretion. To our knowledge, no prior studies have explored the potential role of SPRY2 in cells or tissues relevant to these conditions.
The liver is a central metabolic organ and plays a critical role in lipid metabolism and glucose homeostasis. Hence, we aimed to functionally characterise SPRY2 in HepG2 cells, an in vitro model of human hepatocytes widely studied in the context of glucose and lipid metabolism and insulin resistance [17,18,19]. We observed a marked increase in glucose uptake, along with an increase in lipid droplet accumulation in HepG2 cells after knockout of SPRY2. Transcriptomic profiling revealed differentially expressed genes related to cholesterol biosynthesis, regulation of cell cycle and cellular signalling. These findings suggest a role for SPRY2 in hepatocyte metabolism and provide further evidence that SPRY2 is the likely causal gene in a well-established locus associated with body fat distribution and T2DM.
Methods
Cell culture
Human hepatoma HepG2 cells (ATCC, HB-8065) were cultured in DMEM + GlutaMAX (Gibco; containing 1 g/L glucose) supplemented with 10% foetal bovine serum (FBS), 100 units/mL penicillin, 0.1 mg/mL streptomycin (all Gibco) and 5 μg/mL plasmocin (Invivogen). Cells were serum-starved overnight prior to assays.
CRISPR-Cas9 genome editing
Single guide RNAs (sgRNA) targeting two distinct regions of the human SPRY2 gene were designed using the online tool at: www.broadinstitute.org/gpp/public/analysis-tools/sgrna-design (Additional file 1: Figure S2) and cloned into the BsmBI site of the lentiCRISPRv2 lentiviral vector (Feng Zhang; Addgene #52961) according to [20]. The sgRNA sequences were: 5′-AGTCTCACTGTTGTACACGAtgg-3′ and 5′-GGTTGCCTTAAATTGTGCCAggg-3′ (PAM sequences shown in lower case letters). Correct insertion was verified by Sanger sequencing. Lentiviruses expressing Cas9 and the sgRNA were generated in HEK293T cells by co-transfection of the packaging plasmids psPAX.2 (Didier Trono; Addgene #12260) and psMD2 (David Ron; Addgene #21799). Supernatants containing lentivirus were harvested 24 h and 48 h post-transfection. The pLJM1-EGFP plasmid (David Sabatini; Addgene #19319) was used as a transduction control. HepG2 cells were transduced in OptiMEM (Gibco) containing 8 μg/mL hexadimethrine bromide (polybrene; Sigma-Aldrich) and LentiBlast Reagent (OZ Biosciences). Mock (control) and selection control cells received OptiMEM in place of lentivirus. Transduced cells were selected with puromycin (Gibco) at a concentration of 1 μg/mL for 5–7 days.
Assessment of CRISPR-Cas9 editing efficiency
Assessment of CRISPR-Cas9 genome editing efficiency for SPRY2 was determined by sequencing of isolated alleles of the targeted region as described in Additional file 1. Independent transductions were assessed for efficiency of genome editing using Tracking of Indels by Decomposition (TIDE) [21] (Additional file 1: Figure S3). Confirmation of SPRY2 genome editing was also determined at the protein and mRNA level.
Overexpression of SPRY2
The coding sequence of the SPRY2 gene was amplified from human cDNA using the following primers: 5′-ccgctagccaccATGGAGGCCAGAGCTCAGAGTGGC-3′ and 5′-ccttcgaaCTATGTTGGTTTTTCAAAGTTCC-3′ (start and stop codons are shown in bold capital letters; restriction sites used for cloning are shown in bold small letters). Amplification was carried out using Q5 High-Fidelity DNA Polymerase (New England Biolabs) with the following PCR conditions: 30 s at 98 °C, followed by two touchdown cycles of: 7 s at 98 °C, 20 s at 60 °C, 59 °C, 58 °C and 70 s at 72 °C and ending with 26 cycles of: 7 s at 98 °C, 20 s at 57 °C and 70 s at 72 °C and a final extension of 2 min at 72 °C. The expected 968 bp DNA band was isolated, digested with NheI/BstB1 and cloned in the lentiviral vector pLJM1-EGFP that had previously been cut with NheI/BstB1, excising the coding EGFP sequence. The obtained vector (pLJM1-SPRY2) was sequenced before transduction was performed as described above.
Glucose uptake assay
Glucose uptake was measured using the fluorescent glucose analogue, 2-NBDG (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose; Thermo Fisher) [22]. Cells were cultured and differentiated in black-sided 96-well plates. On the day of the assay, cells were glucose- and serum-starved for 45 min, then 100 μg/mL 2-NBDG in glucose- and serum-free medium was applied for 30 min. Cells were rinsed twice in PBS and a solution of 3.3 μM Hoechst 33342 (Thermo Fisher) in PBS was applied to each well. Cells were imaged with the EVOS FL Auto Imaging System (Life Technologies) on the GFP and DAPI channels at 10x magnification (9–16 images per well). Images were analysed in CellProfiler v. 2.2.0 [23] using an automated pipeline created in-house to subtract background fluorescence, measure GFP intensity and count the number of nuclei in each image. Mean GFP fluorescence per cell, representing glucose uptake, was then calculated.
Lipid droplet accumulation
The fluorescent neutral lipid dye, Bodipy 493/503 (4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene; Molecular Probes), was used to measure the accumulation of lipid droplets in HepG2 cells, as an alternative to Oil Red O [24]. A stock solution of Bodipy 493/503 was prepared in ethanol at a concentration of 1 mg/mL. Cells were treated with 0.5 μg/mL Bodipy 493/503 and 3.3 μM Hoechst 33342 in PBS and imaged with the EVOS FL Auto Imaging System on the GFP and DAPI channels at 10x magnification (9–16 images per well). Images were analysed in CellProfiler as described for the glucose uptake assay.
Phospho-kinase Array
Considering the established role of SPRY2 in RTK signalling [11, 25], we hypothesised that key cellular signalling pathways may be affected in these cells. To investigate this, we performed a phospho-kinase array for the simultaneous determination of phosphorylation levels in 43 protein kinases across multiple pathways relevant to metabolism and insulin resistance (including Akt, MAPK, mTOR and Jak/STAT signalling). This was carried out using the Proteome Profiler Human Phospho-Kinase Array (R&D Systems) following the manufacturer’s protocol for LI-COR near-infrared fluorescence detection. Cells were incubated in serum-free medium with or without insulin (100 nM for 10 min [19]) then lysed in Lysis Buffer 6 from the array kit. Protein concentration was determined using the bicinchoninic acid (BCA) assay (Thermo Fisher). Total cellular proteins (300 μg) were hybridised to the array membranes overnight. The arrays were washed and incubated with the detection antibody cocktails for 2 h, followed by IRDye 800CW Streptavidin (LI-COR 925–32,230; 1:2000) for 30 min. Membranes were imaged on the LI-COR Odyssey infrared imaging system and density of spots determined using Fiji [26].
RNA extraction, library preparation and RNA sequencing (RNA-Seq)
RNA was extracted from mock and SPRY2 KO HepG2 cells (four preparations each) and sequenced on the Ion Proton System using the Ion PI Hi-Q Sequencing 200 Kit (Thermo Fisher). Full details are provided in Additional file 1.
Analysis of RNA-Seq data
Differential expression of genes in mock and SPRY2 KO samples was assessed using the DESeq2 R package from Bioconductor (http://bioconductor.org/packages/release/bioc/html/DESeq2.html). Enrichment of functional terms: gene ontology (GO) molecular process, GO biological process and GO cellular component, PANTHER pathways [27] and Reactome pathways [28] was performed with the AmiGO 2 tool. Analysis methods are described in detail in Additional file 1.
Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
RNA extraction, reverse transcription and real-time PCR were carried out as outlined in Additional file 1. Primer sequences for genes of interest identified by RNA-Seq and the candidate reference genes RPL13, HPRT and B2M are shown in Additional file 1: Table S1. qPCR data analysis was carried out with qbase+ software, version 3.1 (Biogazelle; www.qbaseplus.com), which included a geNorm reference gene study to determine the most stable reference genes for accurate data normalisation.
Statistical analysis
All results are presented as mean ± SD. Raw fluorescence data were combined and corrected according to the method described in [29] to eliminate additive and multiplicative systematic errors. Statistical analyses were performed using GraphPad Prism version 5.00 for Windows, GraphPad Software, www.graphpad.com, with P < 0.05 considered significant. Details of specific statistical tests are shown in the Figure legends.
Results
Confirmation of successful CRISPR-Cas9 genome editing
For the SPRY2 KO studies, HepG2 cells were transduced with two distinct CRISPR-Cas9 sgRNA-lentiviral constructs to minimise the risk of off-target effects, which can compromise specificity [30]. The resultant transduced cultures are referred to as SPRY2 KO1 and SPRY2 KO2. Western blot analyses showed SPRY2 protein expression to be reduced to 30 ± 4% of mock levels in the SPRY2 KO1 cells, and 15 ± 1% in the SPRY2 KO2 cells (mean ± SD of 2 independent transductions; Fig. 1a-b). For the SPRY2 overexpression (OE) study, transduction of HepG2 cells with the pLJM1-SPRY2 lentiviral vector led to a 4-fold increase in SPRY2 band density compared to mock (Fig. 1c-d).

Confirmation of CRISPR-Cas9 genome editing for SPRY2. Near-infrared Western blotting was carried out to simultaneously detect SPRY2 and β-tubulin in HepG2 untreated (mock) and CRISPR-Cas9 genome edited (KO/OE) cell lysates. SPRY2 band density was quantified in Fiji, normalised to β-tubulin and expressed as a percentage of mock. a Representative Western blot for KO study and b band density analysis showing mean + SD of 2 independent transductions. ‘GFP’ denotes transduction control cells expressing GFP using the pLJM1-EGFP lentiviral vector. c Western blot for SPRY2 overexpression (OE) study and d corresponding band density analysis. Full-length blots are shown in Additional file 1: Figure S8
SPRY2 KO increases glucose uptake and lipid droplet accumulation in HepG2 cells
The fluorescent glucose analogue, 2-NBDG, was used to measure glucose uptake in HepG2 mock and SPRY2 KO cells. Knockout of SPRY2 resulted in significantly higher glucose uptake compared to mock (Fig. 2a; Additional file 1: Figure S4). Glucose uptake was increased by (fold changes relative to mock): 1.4 ± 0.1 in the SPRY2 KO1 cells and 1.4 ± 0.2 in the SPRY2 KO2 cells (P < 0.05). There were no differences in glucose uptake between HepG2 mock cells and those overexpressing SPRY2.

SPRY2 KO increases glucose uptake and lipid droplet accumulation in HepG2 cells. a HepG2 mock, SPRY2 KO and SPRY2 OE cells were treated with 100 μg/mL 2-NBDG for 30 min. Cells were washed in PBS, treated with 3.3 μM Hoechst 33342 in PBS and imaged on the GFP and DAPI channels of the EVOS fluorescence microscope. The mean fluorescence per cell, representing glucose uptake, was calculated using CellProfiler and expressed relative to mock. Bars depict mean + SD from 3 independent experiments, with 2 to 4 wells per condition, 9–16 images per well. *P < 0.05 by one-way ANOVA with Bonferroni’s post-hoc test comparing KO with mock. b Cells were treated with Bodipy 493/503 (0.5 μg/mL) to label intracellular lipids and imaged using fluorescence microscopy. CellProfiler was used to calculate the mean GFP fluorescence per cell, representing lipid droplet accumulation. Bars depict mean + SD from 3 independent experiments, relative to mock, with 2 to 4 wells per condition, 9–16 images per well. **P < 0.01, ***P < 0.001 by one-way ANOVA with Bonferroni’s post-hoc test; ‘ns’ denotes not significant
To measure lipid droplet accumulation, the neutral lipid stain, Bodipy 493/503, was applied and visualised with fluorescence microscopy. Lipid droplet accumulation was significantly higher in SPRY2 KO1 and KO2 cells vs. mock (fold changes relative to mock: 1.5 ± 0.1 for SPRY2 KO1, P < 0.01; 1.9 ± 0.04 for SPRY2 KO2, P < 0.001). There were no differences in lipid droplet accumulation between SPRY2 OE and mock cells (Fig. 2b; Additional file 1: Figure S5).
Extent of protein kinase phosphorylation following SPRY2 KO
Next, the phosphorylation of 43 protein kinases was simultaneously investigated using a phospho-kinase array. Untreated and insulin-stimulated mock and SPRY2 KO2 cell lysates were evaluated for each cell type; SPRY2 KO2 cultures were selected since they displayed more compelling phenotypes than SPRY2 KO1 in the metabolic assays. However, there were no significant changes in any of the 43 phosphorylated protein kinases between mock and KO cells, based on our a priori defined requirement of ≥2.0-fold difference (Additional file 1: Figure S6).
SPRY2 KO impacts transcript levels across multiple metabolic pathways
We investigated downstream effects of SPRY2 KO at the transcriptional level in HepG2 cells using RNA-Seq (Figs. 3 and 4; Additional file 1: Figure S7). Analysis of differential expression using DESeq2 revealed 178 differentially expressed (DE) upregulated genes and 243 DE downregulated genes (FDR < 0.1, fold changes ≥2.0) when comparing SPRY2 KO2 to mock RNA samples (Fig. 3; Additional file 2).

Effect of SPRY2 KO on transcript levels in HepG2 cells by RNA-Seq. a Volcano plot depicting up- and downregulated genes (green) resulting from SPRY2 KO in HepG2 cells. b Principal component analysis (PCA) showing separation of mock and SPRY2 KO samples along the major PC1 axis. c Clustering of the top 100 differentially expressed genes indicating separation of mock and SPRY2 KO samples. d MA plot showing relationship between P value and fold change; DE genes are labelled in red

Enrichment analysis of DE genes following SPRY2 KO in HepG2 cells. Top GO Biological Process terms identified in a downregulated and b upregulated DE genes from RNA-Seq analysis. The value of -log10 P reflects the significance of the GO term enrichment (Enr)
The DE genes were subsequently tested for the enrichment of GO biological process, GO molecular process, GO cellular component, PANTHER and Reactome pathways in order to classify them by general biological function and pathway involved. Within GO biological processes (Fig. 4), the upregulated DE genes showed enrichment over the expected number of genes per category for mitotic DNA replication (51-fold), cell cycle regulation (G1/S transition: 10-fold, metaphase/anaphase: 13-fold), plasma lipoprotein particle remodelling (17-fold), regulation of cholesterol biosynthetic process (14-fold) and unsaturated fatty acid biosynthetic process (14-fold). In the set of downregulated DE genes, the categories were more diverse and included cellular responses to metal ion (zinc: 22-fold, cadmium: 19-fold, calcium: 6-fold) and regulation of toll-like receptor (TLR) signalling pathways (14-fold), among others. Pathway analysis results are summarised in Additional file 3. Notably, within GO molecular processes, upregulated genes were enriched in low (23-fold) and very low density lipoprotein (VLDL; 90-fold) particle receptor binding. Within the GO cellular component, upregulated genes were enriched for high-density lipoprotein particle (18-fold), while the group of downregulated genes showed 53-fold enrichment of the inhibitor of κB (I-κB)/nuclear factor κB (NF-κB) complex. Upregulated Reactome pathways were mainly related to cell cycle, but also to lipid digestion, mobilisation and transport (7-fold), VLDL interactions (16-fold) and regulation of cholesterol biosynthesis by SREBF (9-fold).
Validation of RNA-Seq results
The top 20 differentially expressed genes identified by RNA-Seq that were obtained using DE analysis are shown in Table 1. PLA2G2A (secretory phospholipase A2 group IIA) was the most significant upregulated gene (fold change 3.9; adjusted P = 1.43E− 20), which is of particular interest to metabolic diseases in the context of atherogenesis [31], hepatic cholesterol metabolism [32] and insulin sensitivity [33]. The most significant downregulated gene was GADD45B (growth arrest and DNA damage inducible beta; fold change 0.2; adjusted P = 2.00E− 20), which is involved in cellular signalling events related to cell survival, DNA repair and apoptosis [34]. There were several other notable DE genes within the top 20 list with relevance to obesity and T2DM, including: DPP4 (dipeptidyl peptidase 4) [35], TNFAIP3 (tumour necrosis factor alpha inducible protein 3), CYP1A1 (cytochrome P450 family 1 subfamily A member 1) and AKR1B1 (aldo-keto reductase family 1 member B). Finally, nominal significance was observed for genes involved in glucose transport, fatty acid synthesis and glycolysis, including sterol regulatory element-binding protein 1 (SREBF1) and glucokinase regulator (GCKR) (Additional file 1: Figure S7).
We subsequently carried out RT-qPCR experiments to validate selected genes of interest from the top 20 list of DE genes in HepG2 mock, GFP and SPRY2 KO cells (Fig. 5). PLA2G2A mRNA level was confirmed to be significantly upregulated in both SPRY2 KO1 and KO2 cells (fold changes relative to mock: 2.7 ± 0.5 for SPRY2 KO1, P < 0.05; 4.7 ± 1.1 for SPRY2 KO2, P < 0.001). There was a significant decrease in DPP4 mRNA level in the SPRY2 KO2 cells (0.6 ± 0.06 relative to mock, P < 0.05). There were no differences in TNFAIP3 mRNA level between any of the samples. Finally, significant changes were detected in the SPRY2 KO samples relative to mock (but not GFP) in the following genes: GADD45B, CYP1A1 and AKR1B1.

Validation of RNA-Seq results by RT-qPCR. Relative mRNA level in HepG2 mock, GFP and SPRY2 KO cells as assessed by RT-qPCR, for: a PLA2G2A, b DPP4, c GADD45B, d TNFAIP3, e CYP1A1 and f AKR1B1. Data were analysed in qbase+ and normalised to HPRT and RPL13, determined by geNorm to be the most stable reference genes. Bars show mean + SD from 3 biological replicates each, quantified in triplicate and expressed relative to mock. *P < 0.05, **P < 0.01, ***P < 0.001 by one-way ANOVA with Dunnett’s post-hoc test to compare all bars to mock
Discussion
To our knowledge, this is the first study to functionally characterise SPRY2, a gene highlighted by GWAS for BF% [6] and T2DM [8], in human hepatocytes. Our results show increased glucose uptake and elevated lipid droplet accumulation to be the major functional consequences of SPRY2 KO in these cells, as well as modifications to transcript levels of several genes and biological pathways relevant to the pathogenesis of metabolic diseases, such as obesity and T2DM.
The liver is a central metabolic organ and plays a critical role in lipid metabolism and glucose homeostasis. These processes can be dysregulated in obesity, leading to metabolic abnormalities that are associated with the development of NAFLD, insulin resistance and the pathogenesis of T2DM [36]. HepG2 cells are widely used for the study of glucose and lipid metabolism, insulin signalling and mechanisms of insulin resistance in vitro [17,18,19], and were therefore selected as a model of human hepatocytes in the present study.
The observed significant increase in glucose uptake following SPRY2 KO in HepG2 cells suggests a possible role for SPRY2 in glucose metabolism in hepatocytes. However, the mechanism by which this enhanced glucose uptake occurs is unclear; our RNA-Seq analysis revealed a nominal reduction in transcript levels of SLC2A1 (GLUT1, the major glucose transporter in HepG2 cells [37]) in SPRY2 KO cells compared to mock.
Given the association of the rs534870 variant near SPRY2 with BF% [6], as well as its postulated specific effect on adiposity [7], we hypothesised that SPRY2 KO could affect lipid droplet accumulation in HepG2 cells. Indeed, there were significantly more lipid droplet in the SPRY2 KO cells compared to mock; this may be a result of de novo fatty acid synthesis due to increased glucose uptake [38]. Our RNA-Seq data showed that SPRY2 KO led to an increase in transcript level of enzymes involved in lipogenesis, including acetyl-CoA carboxylase alpha (ACACA), the rate-limiting step in long-chain fatty acid synthesis [39], as well as fatty acid synthase (FASN). Furthermore, SPRY2 KO may enhance the glycolytic pathway that provides pyruvate for fatty acid synthesis: our RNA-Seq results showed decreased transcript levels of GCKR, which negatively regulates hepatic glucokinase (GCK), a glycolytic enzyme that phosphorylates glucose to produce glucose-6-phosphate [40], as well as a significant increase in transcript level of SREBF1, a transcription factor that mediates insulin-stimulated upregulation of GCK [41]. Therefore, SPRY2 KO may affect glucose uptake and lipid droplet accumulation in part by modulating activation and expression of insulin-sensitive GCK enzymes, although these observations require validation in future studies.
A clear phenotype resulting from SPRY2 OE in HepG2 cells did not emerge in the present study, with similar levels of glucose uptake and lipid droplet accumulation observed in SPRY2 OE and mock cells. Gain- and loss-of-function studies of a target gene are generally expected to produce opposite phenotypes, however, this is not always observed [42, 43], particularly with regard to tumour suppressor genes [44], which may apply in the case of SPRY2. Ideally, the SPRY2 OE experiments would be repeated to further characterise the resultant phenotypes in HepG2 cells.
Further investigation into cellular signalling pathways via phospho-kinase profiling did not reveal any significant changes to protein kinase phosphorylation levels between HepG2 mock and SPRY2 KO cells, despite the array encompassing a number of protein kinases relevant to glucose metabolism and insulin signalling. Transcriptome profiling via RNA-Seq identified a total of 421 DE genes between mock and SPRY2 KO HepG2 cells (178 upregulated and 243 downregulated genes). The most significant DE gene, PLA2G2A, was validated by RT-qPCR as significantly upregulated at the mRNA level following SPRY2 KO. PLA2G2A is highly expressed in liver and elevated circulating levels of its encoded protein, secretory phospholipase A2 group IIA (sPLA2), are a risk factor for atherogenesis [31]. Furthermore, sPLA2 influences hepatic cholesterol uptake [32] and improves insulin sensitivity in mice [33]. Dipeptidyl peptidase 4 (DPP4) mRNA level was validated as significantly decreased in the SPRY2 KO2 cells. Elevated DPP4 expression has been linked to insulin resistance in obesity [45] and NAFLD [35], and DPP4 inhibitors are currently in clinical use as anti-diabetic drugs. Further studies will be required to explore the potential link between SPRY2 and these genes in the context of obesity and T2DM, and in particular, the potential role of SPRY2 in the pathogenesis of liver diseases, such as NAFLD.
Pathway analysis revealed many DE upregulated genes arising from SPRY2 KO to be involved with DNA replication and cell cycle regulation; this is consistent with the established role for SPRY2 in inhibiting cell proliferation and acting as a tumour suppressor in certain types of cancer [14, 46]. Among the highly significant terms several were related to the metabolic processes that regulate cholesterol biosynthesis and fatty acid metabolism, which correlates with our experimental findings suggesting that SPRY2 regulates metabolic processes, such as glucose uptake and lipid accumulation.
Collectively, the increase in glucose uptake and lipid droplet accumulation, as well as the modulation of transcript levels in SPRY2 KO cells suggest that SPRY2 may be involved in metabolic homeostasis in hepatocytes, although whether this contributes to the pathogenesis of obesity, T2DM or NAFLD remains to be determined using further in vitro experiments. However, it would be interesting to speculate that the changes in transcript levels of multiple genes associated with fatty acid synthesis and alterations to cholesterol synthesis pathways observed in the SPRY2 KO cells could constitute a potential mechanism leading to increased lipid droplet accumulation in hepatocytes, and potentially, a contribution by SPRY2 to conditions such as hepatic steatosis and NAFLD. Furthermore, the present study is unable to disentangle if the numerous DE cell cycle regulation genes identified in the RNA-Seq experiments are relevant to obesity and T2DM or may also be indicative of neoplastic changes, given that the loss of SPRY2 is associated with hepatocarcinogenesis [47,48,49] and that HepG2 cells themselves are a hepatoma cell line.
Conclusions
We have shown that SPRY2 KO increases glucose uptake and lipid droplet accumulation in HepG2 cells, and leads to downstream transcriptomic changes in genes relevant to metabolic diseases. Although the present study does not identify the exact molecular mechanisms underlying the observed metabolic changes in the SPRY2 KO cells, it provides important insights into the function of SPRY2 in human hepatocytes and contributes to elucidating the potential role of SPRY2 in the pathogenesis of obesity and T2DM.
Availability of data and materials
The data generated or analysed during this study are included in this published article (and its supplementary information files).
Abbreviations
- BF%:
-
Body fat percentage
- BMI:
-
Body mass index
- DE:
-
Differentially expressed
- GWAS:
-
Genome-wide association studies
- KO:
-
Knockout
- NAFLD:
-
Non-alcoholic fatty liver disease
- RTK:
-
Receptor tyrosine kinase
- sgRNA:
-
single guide RNA
- SPRY2:
-
Sprouty RTK signalling antagonist 2
- T2DM:
-
Type 2 diabetes mellitus
References
Chooi YC, Ding C, Magkos F. The epidemiology of obesity. Metabolism. 2019;92:6–10.
Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. N Engl J Med. 2017;376(3):254–66.
Kovesdy CP, L., Furth S, Zoccali C, on behalf of the World Kidney Day Steering Committee. Obesity and kidney disease: hidden consequences of the epidemic. Clin Kidney J. 2017;10(1):1–8.
GBD 2015 Obesity Collaborators, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N Engl J Med. 2017;377(1):13–27.
Ghosh S, Bouchard C. Convergence between biological, behavioural and genetic determinants of obesity. Nat Rev Genet. 2017;18(12):731–48.
Kilpelainen TO, Zillikens MC, Stancakova A, Finucane FM, Ried JS, Langenberg C, et al. Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet. 2011;43(8):753–60.
Lu Y, Day FR, Gustafsson S, Buchkovich ML, Na J, Bataille V, et al. New loci for body fat percentage reveal link between adiposity and cardiometabolic disease risk. Nat Commun. 2016;7:10495.
DIAbetes Genetics Replication Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES) Consortium, Mahajan A, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234.
Shu XO, Long J, Cai Q, Qi L, Xiang YB, Cho YS, et al. Identification of new genetic risk variants for type 2 diabetes. PLoS Genet. 2010;6(9):e1001127.
Imamura M, Iwata M, Maegawa H, Watada H, Hirose H, Tanaka Y, et al. Genetic variants at CDC123/CAMK1D and SPRY2 are associated with susceptibility to type 2 diabetes in the Japanese population. Diabetologia. 2011;54(12):3071–7.
Impagnatiello MA, Weitzer S, Gannon G, Compagni A, Cotten M, Christofori G. Mammalian sprouty-1 and -2 are membrane-anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J Cell Biol. 2001;152(5):1087–98.
Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feed back regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16(1):45–54.
Yusoff P, Lao DH, Ong SH, Wong ESM, Lim J, Lo TL, et al. Sprouty2 inhibits the Ras/MAP kinase pathway by inhibiting the activation of raf. J Biol Chem. 2002;277(5):3195–201.
Gross I, Bassit B, Benezra M, Licht JD. Mammalian Sprouty proteins inhibit cell growth and differentiation by preventing Ras activation. J Biol Chem. 2001;276(49):46460–8.
Masoumi-Moghaddam S, Amini A, Morris DL. The developing story of Sprouty and cancer. Cancer Metastasis Rev. 2014;33(2–3):695–720.
Pappalardo Z, Gambhir Chopra D, Hennings TG, Richards H, Choe J, Yang K, et al. A whole-genome RNA interference screen reveals a role for Spry2 in insulin transcription and the unfolded protein response. Diabetes. 2017;66(6):1703–12.
Zang M, Zuccollo A, Hou X, Nagata D, Walsh K, Herscovitz H, et al. AMP-activated protein kinase is required for the lipid-lowering effect of metformin in insulin-resistant human HepG2 cells. J Biol Chem. 2004;279(46):47898–905.
Ishii M, Maeda A, Tani S, Akagawa M. Palmitate induces insulin resistance in human HepG2 hepatocytes by enhancing ubiquitination and proteasomal degradation of key insulin signaling molecules. Arch Biochem Biophys. 2015;566:26–35.
Cordero-Herrera I, Martin MA, Goya L, Ramos S. Cocoa flavonoids attenuate high glucose-induced insulin signalling blockade and modulate glucose uptake and production in human HepG2 cells. Food Chem Toxicol. 2014;64:10–9.
Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–7.
Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42(22):e168.
Zou C, Wang Y, Shen Z. 2-NBDG as a fluorescent indicator for direct glucose uptake measurement. J Biochem Biophys Methods. 2005;64(3):207–15.
Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7(10):R100.
Spangenburg EE, Pratt SJ, Wohlers LM, Lovering RM. Use of BODIPY (493/503) to visualize intramuscular lipid droplets in skeletal muscle. J Biomed Biotechnol. 2011;2011:598358.
Hanafusa H, Torii S, Yasunaga T, Nishida E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat Cell Biol. 2002;4(11):850–8.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–82.
Mi H, Muruganujan A, Ebert D, Huang X, Thomas P. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2019;47(D1):D419–D26.
Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2018;46(D1):D649–D55.
Echenique-Robba P, Nelo-Bazan MA, Carrodeguas JA. Reducing the standard deviation in multiple-assay experiments where the variation matters but the absolute value does not. PLoS One. 2013;8(10):e78205.
Zhang XH, Tee LY, Wang XG, Huang QS, Yang SH. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther Nucleic Acids. 2015;4:e264.
Kugiyama K, Ota Y, Takazoe K, Moriyama Y, Kawano H, Miyao Y, et al. Circulating levels of secretory type II phospholipase a (2) predict coronary events in patients with coronary artery disease. Circulation. 1999;100(12):1280–4.
Eckey R, Menschikowski M, Lattke P, Jaross W. Increased hepatic cholesterol accumulation in transgenic mice overexpressing human secretory phospholipase a (2) group IIA. Inflammation. 2004;28(2):59–65.
Kuefner MS, Pham K, Redd JR, Stephenson EJ, Harvey I, Deng X, et al. Secretory phospholipase a (2) group IIA modulates insulin sensitivity and metabolism. J Lipid Res. 2017;58(9):1822–33.
Niehrs C, Schafer A. Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol. 2012;22(4):220–7.
Baumeier C, Schluter L, Saussenthaler S, Laeger T, Rodiger M, Alaze SA, et al. Elevated hepatic DPP4 activity promotes insulin resistance and non-alcoholic fatty liver disease. Mol Metab. 2017;6(10):1254–63.
Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56(4):952–64.
Karim S, Adams DH, Lalor PF. Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol. 2012;18(46):6771–81.
Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism. 2014;63(7):895–902.
Tong L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cell Mol Life Sci. 2005;62(16):1784–803.
Gidh-Jain M, Takeda J, Xu LZ, Lange AJ, Vionnet N, Stoffel M, et al. Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships. Proc Natl Acad Sci U S A. 1993;90(5):1932–6.
Massa ML, Gagliardino JJ, Francini F. Liver glucokinase: an overview on the regulatory mechanisms of its activity. IUBMB Life. 2011;63(1):1–6.
Li ZJ, Chen P, Su R, Li YY, Hu C, Wang YG, et al. Overexpression and knockout of miR-126 both promote leukemogenesis. Blood. 2015;126(17):2005–15.
Prelich G. Gene overexpression: uses, mechanisms, and interpretation. Genetics. 2012;190(3):841–54.
Wang PP, Zhou ZH, Hu AC, de Albuquerque CP, Zhou Y, Hong LX, et al. Both decreased and increased SRPK1 levels promote Cancer by interfering with PHLPP- mediated Dephosphorylation of Akt. Mol Cell. 2014;54(3):378–91.
Ghorpade DS, Ozcan L, Zheng Z, Nicoloro SM, Shen Y, Chen E, et al. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature. 2018;555(7698):673–7.
Zhang Q, Shim K, Wright K, Jurkevich A, Khare S. Atypical role of sprouty in p21 dependent inhibition of cell proliferation in colorectal cancer. Mol Carcinog. 2016;55(9):1355–68.
Fong CW, Chua M-S, McKie AB, Ling SHM, Mason V, Li R, et al. Sprouty 2, an inhibitor of mitogen-activated protein kinase signaling, is Down-regulated in hepatocellular carcinoma. Cancer Res. 2006;66(4):2048.
Wang C, Delogu S, Ho C, Lee SA, Gui B, Jiang L, et al. Inactivation of Spry2 accelerates AKT-driven hepatocarcinogenesis via activation of MAPK and PKM2 pathways. J Hepatol. 2012;57(3):577–83.
Lee SA, Ladu S, Evert M, Dombrowski F, De Murtas V, Chen X, et al. Synergistic role of Sprouty2 inactivation and c-met up-regulation in mouse and human Hepatocarcinogenesis. Hepatology. 2010;52(2):506–17.
Acknowledgements
The authors acknowledge support from Science for Life Laboratory, the National Genomics Infrastructure, NGI, and UPPMAX for providing assistance in massive parallel sequencing and computational infrastructure.
Funding
The authors gratefully acknowledge support from the Swedish Heart-Lung Foundation (grant no. 20140422), the Swedish Research Council (grant no. 2015–02907), Knut och Alice Wallenberg Foundation (no. 2013.0126) and National Institutes of Health (NIH; 1R01 DK106236-01A1, R01DK107437). The funding bodies did not play any part in the design of the study, collection, analysis and interpretation of data or in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
NLC designed and carried out experiments, analysed data and drafted the manuscript. MP analysed the RNA-Seq data and helped write the manuscript. AGE designed computational analysis pipelines and analysed data. ASR assisted with RNA-Seq analysis. SH contributed to methodology and interpretation. JWK and TQ contributed to design and interpretation. CCL designed and carried out molecular biology experiments, contributed to data analysis and helped direct the project. EI devised and directed the project, contributed to study design and supervision, and revised the manuscript. All authors discussed the results, contributed to and have approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Supplementary Methods. Figure S1. Overview of the SPRY2 locus. Figure S2. Exon organisation of SPRY2 gene. Figure S3. Examples of DNA mutations generated following CRISPR-Cas9 genome editing for SPRY2 in HepG2 cells. Figure S4. SPRY2 KO increases glucose uptake in HepG2 cells. Figure S5. Increased lipid droplet accumulation following SPRY2 KO in HepG2 cells. Figure S6. SPRY2 KO does not alter protein kinase phosphorylation in HepG2 cells. Figure S7. Alterations in the expression of key genes involved in glucose transport, lipogenesis and glycolysis following SPRY2 KO in HepG2 cells. Figure S8. Confirmation of CRISPR-Cas9 genome editing for SPRY2. Table S1. Primer sequences for RT-qPCR.
Additional file 2.
Differential gene expression comparing HepG2 mock and SPRY2 KO cells. Separate tabs for complete list, significantly up- and downregulated transcripts.
Additional file 3.
Pathway analysis of differentially expressed genes identified by RNA-Seq displaying results for enrichment of PANTHER, Reactome and GO biological process sets.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cook, N.L., Pjanic, M., Emmerich, A.G. et al. CRISPR-Cas9-mediated knockout of SPRY2 in human hepatocytes leads to increased glucose uptake and lipid droplet accumulation. BMC Endocr Disord 19, 115 (2019). https://doi.org/10.1186/s12902-019-0442-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12902-019-0442-8