
Abstract
Background
As a main drug for diseased thrombus, some clinically used thrombolytic agents have various disadvantages, safer novel thrombolytic agents are of great demand. This study aimed to achieve high and efficient production of a fibrinolytic enzyme with superior enzymatic properties, by a combination strategy of codon optimization, gene dosage and process optimization in Komagataella phaffii (K. phaffii).
Results
After codon optimization, the fibase from a marine Bacillus subtilis was expressed and secreted in K. phaffii GS115. Recombinant strains harboring different copies of the fib gene (fib-nc) were successfully obtained via Geneticin (0.25–4 mg/ml) screening on minimal dextrose selection plates and assessment via real-time quantitative PCR. The respective levels of fibase produced by strains expressing fib-5.4c, fib-6c, fib-8c, fib-9c, and fib-12c were 4428, 5781, 7323, 7930, and 2472 U/ml. Levels increased as the copy number increased from 4 to 9, but decreased dramatically at copy number 12. After high cell density fermentation optimization, the highest fibase activity of the strain expressing fib-9c was 7930 U/ml in a shake flask and increased to 12,690 U/ml after 3 days of continuous culture in a 5-L fermenter, which is one of the highest levels of production reported. The recombinant fibase was maximally active at pH 9.0 and 45 °C, and was remarkably stable at pH levels ranging from 5 to 10 and temperatures up to 50 °C. As a metal-dependent serine protease, fibase did not cause hemolysis in vitro and preferentially degraded fibrin directly.
Conclusions
The combination of codon optimization, gene dosage, and process optimization described herein could be used for the expression of other therapeutic proteins difficult to express. The characteristics of the recombinant fibase suggest that it has potential applications for thrombosis prevention and therapy.
Similar content being viewed by others

Background
In recent years increasing numbers of cardiovascular diseases caused by thrombosis have begun to contribute to impairment of human health, and they are the main reason for morbidity and mortality worldwide [1]. As the main medical drug for diseased thrombus, thrombolytic agents such as urokinase, streptokinase, and tissue-type plasminogen activator have been widely used in clinical therapy. There have always been associated disadvantages however, including high cost, short half-lives, low fibrin specificity, bleeding complications, and allergic responses [2]. There is therefore a demand for safer novel thrombolytic agents. Many fibrinolytic enzymes (fibases) from animals such as snakes [3], earthworms [4, 5], plants [6], the fungus Cordyceps militaris [7] and microbes [8, 9] have been discovered and studied extensively. However, there are few reports on fibase produced by marine microorganisms.
Microbial fibases are of great interest to researchers due to their potential for use in thrombolytic therapy, which would be of high yield, low cost, and amenable to industrial scale-up. Over-expression of foreign genes in Escherichia coli (E. coli) commonly leads to the formation of inclusion bodies, resulting in inactive proteins. As an expression system, Bacillus subtilis (B. subtilis) has significant advantages over E. coli in that proteins can be readily secreted [10]. But B. subtilis also exhibits high protease expression leading to degradation of target proteins [11], and this high protease expression increases the downstream purification difficulty of target protein. To date two main aspects of protein expression in wild B. subtilis have been investigated. One is the generation of a B. subtilis strain rendered deficient in protease expression via gene knock-out [12, 13]. The other is optimization of fermentation processes with respect to high protein expression. The use of yeasts is conducive to facile genetic manipulation and ease of fermentation of the microorganism, which can result in high yields of proteins in a solution that does not contain pyrogens, pathogens, or viral inclusions [14]. Their rapid growth, microbiological safety, and amenability to high-density fermentation in simple media render them particularly suited to large-scale industrial production of foreign proteins, where secretory expression is important for simplifying the downstream protein purification process [15]. In recent years, as one of the most important industrial organisms for heterologous protein production, Komagataella phaffii (K. phaffii) has been used widely for the production of a broad range of recombinant drug products, including antibody fragments [16], growth factors [17], interferon [18], interleukin [19], insulin precursors [20], and staphylococcal kinase [21] etc.
Despite its high protein productivity, further optimization of K. phaffii expression is imperative due to strain-specific and product-specific challenges such as promoter strength, methanol utilization type, and oxygen demand. To this end, strategies including genetic and process engineering have been employed in this study. Optimizations of codon use and gene dosage have been proved useful for enhancing protein expression levels. Codon optimization includes the adjustment of codon usage bias and GC content, repeat sequence removal, and undesired sequence motif modification. All these factors could render its mRNA secondary structures more stable, increase the translation efficiency and prolong the half-life of mRNA, and finally result in an increase of protein synthesis [22]. Gene dosage (i.e., the RNA abundance at the transcriptional level) is a major limiting factor for high and efficient expression of the targeted gene. Increasing the gene dosage usually has substantial effects on the improvement of protein expression levels [23, 24], and multiple copies of the foreign gene can be inserted at the same site in yeast genomes. Recombinant yeast strains with high expression profiles can be generated by optimizing the number of foreign gene copies inserted. Large-scale production of proteins via high cell density fermentation relies on the optimization of process parameters including methanol feed rate, induction temperature, and specific growth rate [22].
Currently recombinant K. phaffii strains with high copy numbers can only be obtained by increasing gene concatemers, so alternative methods for generating multicopy strains rapidly and reliably are highly desirable [25]. Additionally, in order to study the effects of different promoters on protein expression levels, it is necessary to accurately and quantitatively analyze the copy number of foreign genes inserted into the recombinant yeast. In the process of strain propagation, reduced copy number can indicate plasmid loss. Real-time quantification PCR (qPCR) has been used to detect the copy numbers of many foreign genes including transglutaminase [25], human interleukin-3 [26], and lipase [27] in recombinant yeasts, but to date it has not been used in fib gene copy number estimation.
In the current study, after codon optimization, the fibase from a marine B. subtilis D21 was expressed and secreted in K. phaffii GS115. Multicopy integrants were obtained via screening and qPCR method to enhance the expression of fibase. Shake flask production series were conducted to optimize cultivation parameters such as medium composition, incubation time, temperature, methanol concentration, and coculture with methanol and sorbitol. After high cell density fermentation optimization, the highest fibase activity of the strain expressing fib-9c was 7930 U/ml in a shake flask and increased to 12,690 U/ml after 3 days of continuous culture in a 5-L fermenter, which is one of the highest levels of production reported. Then fibase was purified using one-step Ni-NTA and its biological characteristics was evaluated.
Methods
Strains
The strains and plasmids used are shown in Table 1. E. coli was cultivated in Luria-Bertani medium at 37 °C, and K. phaffii was cultured in buffered glycerol-complex medium (BMGY, 1% yeast extract, 2% peptone, 100 mM potassium phosphate buffer, 4 × 10− 5% biotin, 1.34% Yeast Nitrogen Base, 1% glycerol, pH 6.0) for growth, buffered methanol-complex medium (BMMY, 1% yeast extract, 2% peptone, 100 mM potassium phosphate buffer, 4 × 10− 5% biotin, 1.34% Yeast Nitrogen Base, 0.5% methanol, pH 6.0) for fibase induction, or yeast extract peptone dextrose (YPD) free of glucose (1% yeast extract and 2% peptone) for growth and induction.
Construction, transformation, and screening for recombinant K. phaffii
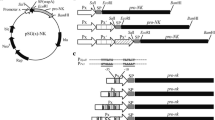
The fib gene (GenBank: KM519994.1) used in this study was derived from a marine B. subtilis D21 screened previously. For high production, 168 codons in total (approximately 61.1%) were replaced with the K. phaffii-preferred codons (Fig. S1). The codon adaptation index was increased from 0.64 of initial sequence to 0.96 of optimized sequence (a codon adaptation index of 0.8–1.0 is regarded as good for high expression). The optimized fib fragment was digested by EcoRI/notI and then cloned into the simultaneously digested pPIC9K vector, together with the a-factor signal peptide under control of the AOX1 promoter. The recombinant plasmid pPIC9K-fib was introduced into competent E. coli DH5α by chemical transformation, positive clones were selected by ampicillin (Amp, 100 μg/ml) resistance, colony PCR and sequencing. The recombinant vector pPIC9K-fib was linearized with SacI and transformed into competent K. phaffii GS115 cells by electroporation, using the set program with a voltage of 1500 V, 25 μF capacitance, and 200 Ω resistance. The construction of recombinant pPIC9K-fib is depicted in Fig. 1a, and the process of integration into the genome is depicted in Fig. 1b. The single copy or high copy recombinant K. phaffii GS115/pPIK9K-fib were selected using minimal dextrose selection plates containing different concentrations of Geneticin (G418, 0.25 mg/ml of 1 copy, 0.5 mg/ml of 1–2 copy, 4 mg/ml of 7–12 copy), colony PCR and sequencing, which were inoculated into glucose-free YPD or BMGY at 30 °C 200 r/min for 24 h, then transferred to BMMY for induce expression.

Construction of recombinant plasmid pPIC9K- fib and its integration sites in the genome
Analysis of fib copy number by real-time quantification PCR
Five positive clones with different fibase activities were selected for analysis of fib copy numbers, which were estimated via qPCR in the K. phaffii genome. The primers and probes used for plasmid construction and qPCR are as shown in Table 2. There is only a single copy of the housekeeper gene TDH1 in the K. phaffii genome [28], so the TDH1 copy number can be used to represent the initial copy number of the genome in the template. After the recombinant plasmid Topo-fib and pMD19T-TDH1 were respectively linearized with XbaI and BamHI as standards, the absolute copy number of the target gene and reference gene were calculated using Eq. 1 below. The standard curve was constructed using real-time fluorescence quantitative Ct values and the logarithm of the copy number of the standard (copies/μl) after gradient dilution. Using genomes with the highest fibase activity and the aforementioned other four positive clones as sample templates, the absolute copy numbers of the target and reference genes were calculated from Ct according to the standard curve, then the copy number of the target gene was calculated using Eq. 2 below.
Fermentation optimization in flasks and 5-L fermenter
In order to establish an efficient and low-cost process for recombinant fibase production, the medium (glucose-free YPD and BMMY), time-course (1–9 days), temperature (28 °C, 32 °C, and 36 °C), methanol concentration for induction (0.5, 1, and 2.5%), and coculture of methanol and sorbitol (0, 2, 4, and 6 g/l) conditions were optimized via an extensive series of shake flask experiments. The shake flask study under optimized condition was performed in 50 ml media in 250 ml shaker flasks. Methanol was added every 24 h to different final concentration to maintain induction. The results of optimization were then verified in scaled up production in a 5-L fermenter with 2.4 l medium volume. The main parameters utilized during the entire fermentation process are shown in Table S1. All values are means ± SD from three independent experiments.
Determination of fibrinolytic activity and protein concentration
Fibrinolytic activity was assayed via the standard fibrin plate method using urokinase [29], with some modifications. Each 10-ml plate contained 0.1% fibrinogen, 1% agarose, and 10 U thrombin. A total of 5 μl of culture supernatant at various dilutions was added to each well. The plate was incubated for 18 h at 37 °C, and fibrinolytic activity was determined based on the standard curve of urokinase. One enzyme unit is defined as the enzyme amount producing a ΔA275 of 1.0 per ml per minute at 37 °C, pH 7.5, when measuring perchloric acid soluble products from α-casein. All data are presented as the mean ± SD of triplicate determinations. The concentration of recombinant fibase was measured using nucleic acid/protein analyzer.
Purification and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
Utilizing the 6His tag at the C-terminal end the recombinant protein was concentrated using an ultrafilter tube and purified with nickel ion affinity chromatograph resin (Ni-NTA) to obtain a single band. The buffer systems utilized were Ni-50 wash buffer (50 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, pH 8.0), Ni-250 elution buffer (250 mM imidazole, 300 mM NaCl, 50 mM NaH2PO4, pH 8.0), cleaning-in-place buffer (0.5 M NaOH), stripping buffer (50 mM Na3PO4·12H2O, 300 mM NaCl, 100 mM EDTA·Na2), and regeneration buffer (100 mM NiSO4·6H2O). All purification processes were conducted at 4 °C. Purification recovery refers to the ratio of total activity before and after purification.
SDS-PAGE was conducted on a Mini-PROTEAN 3 cell apparatus (Bio-Rad, USA) using the tris-glycine discontinuous system with 5% stacking and 12% resolving gels. An unstained protein marker, ProteinRuler II (TRAN, 12–120 kDa) was used as molecular weight standards. Proteins were stained using the Coomassie Brilliant Blue R-250 method.
Characteristics of recombinant protein
Residual fibase activity under the different treatments was determined via the fibrin plate method. The determination of optimal temperature was performed by measuring fibase activity in 0.02 M phosphate-buffered saline (pH 7.4) from 28 °C to 60 °C. The thermal stability of fibase was determined after incubation in the same buffer and temperatures for 1 h or 6 h.
To determine the optimum pH, enzymatic activity measurements were performed at levels ranging from pH 4.0 to pH 10.0. To investigate the pH stability of the purified enzyme, enzyme samples were diluted to the same concentration with 0.02 M disodium hydrogen phosphate-sodium citrate (pH 4.0–6.0), 0.02 M Tris-HCl (pH 7.0–8.0), and 0.02 M glycine-sodium hydroxide (pH 9.0–10.0). Residual fibase activity was measured after incubation at 37 °C for 18 h.
Salt solutions containing different metal ions were prepared, and purified enzymes were added to 5, 20, and 50 mM salt solutions. The residual fibase activity of each enzyme was determined via fibrin plates. Fibase activity with no salt was set as 100%.
The kinetic parameters Km and Vmax for fibase were calculated using chromogenix as the substrate at concentrations of 0.0, 0.1, 0.3, 0.6, 0.9, and 1.2 mM. The kinetic study was performed at 37 °C and pH 7.4 (phosphate buffer, 0.02 M). Fibase was incubated with substrates at different concentrations at 37 °C for 8 min, and absorption values at 405 nm were measured. The amount (μM) of 4-nitroanilide released by the reaction between tetra-peptide substrate and protease per unit of time was considered to be the velocity of the reaction.
All values above are mean ± SD from three independent experiments.
Results
Expression of yeast codon optimized strains
K. phaffii is a methylotrophic yeast without fibrinolytic activity, the expression efficiency of which is higher than that of Saccharomyces cerevisiae due to the tight regulation and strong inducibility of the AOX1 promoter when methanol is used as the sole carbon source [22]. The recombinant strain GS115 (pPIC9K-fib) was cultivated with a 2% inoculum size and daily addition of 1% (v/v) methanol to a shaking flask containing 50 ml BMMY at pH 6.0 and 30 °C.
A main protein band with a molecular weight of 46 kDa was detected via SDS-PAGE of positive culture supernatants with the highest fibase activity after supernatant concentration (Fig. 2c). No corresponding band was yielded by the transformant with pPIC9k, and the result was consistent with that of fibrin plates (Fig. 2b). Therefore, recombinant fibase was secreted into the supernatant by K. phaffii GS115. The 6His tag at the carbon end facilitated purification of the recombinant protein via nickel ion affinity chromatograph resin, and it was analyzed via SDS-PAGE (Fig. 2a).

Effects of codon optimization on fibase activity and yield. After 5 days of cultivation the culture supernatant was harvested and analyzed via a fibase activity assay (b) and SDS-PAGE (c). The fibase purified via one-step Ni-NTA is 46 kD (a). 1: GS115 (pPIC9K). 2: 125 U/ml urokinase. 3: GS115 (pPIC9K-fib). 4: Purified fibase. M: Protein Ruler II
Quantification of fib copy number in K. phaffii genome
The melt curves of both TDH1 and fib only had a single peak, indicating specificity of the qPCR products (Fig. S2A and S2B). The amplification efficiency of TDH1 was 94.8%, and that of fib was 96.3%. The regression equation of the TDH1 standard curve was y = − 3.441x + 34.952 (R2 = 0.999), and that of fib was y = − 3.511 x + 37.563 (R2 = 0.998) (Fig. 3). The fib copy numbers in the recombinant strain with the highest activity and the other four strains were 5.4, 6.0, 8.0, 8.7 and 12.3 (Table 3). The corresponding strain were named GS115/fib-5.4c, fib-6c, fib-8c, fib-9c, and fib-12c with fibrinolytic activity 4428 U/ml, 5781 U/ml, 7323 U/ml, 7930 U/ml, 2472 U/ml respectively.

Standard curves of the reference gene TDH1 (a) and the target gene fib (b). Data are mean ± SD of three independent runs
Fermentation optimization in flasks and 5-L fermenter
After optimization the maximum fibase activity was obtained with a 2% inoculum size, and the optimal culture conditions included daily addition of 0.5% (v/v) methanol and 2 g/l sorbitol to a 250 ml shaking flask containing 50 ml of glucose-free YPD (1% yeast extract and 2% peptone) culture medium for 120 h at pH 6.0 and 32 °C (Fig. 4). Based on the optimized conditions of fibase recombinant strain cultivation in shaking flasks, the fermentation process in the 5 L-fermenter was established. The highest fibase expression level of the strain expressing fib-9c was 7930 U/ml in a shake flask and increased to 12,690 U/ml after 3 days of continuous culture in a 5-L fermenter (Fig. 5).

Cultivation optimization of recombinant strain GS115/pPIC9K-fib in shaking flasks. The figures show the effect of different media and days (a), induction temperatures (b), methanol concentrations (c), coculture with methanol and sorbitol (d) on enzyme activity (column), growth (line and triangle), and protein concentration (line and circular), respectively. All values are mean ± SD from three independent experiments

The growth, enzyme activity, and protein concentration from fermentations of recombinant strain GS115/pPIC9K-fib in 5-L fermenter. Data are presented as mean ± SD of triplicate experiments
Characteristics of recombinant protein
Effects of pH and temperature on fibase activity and stability.
As shown in Fig. 6, the purified fibase had high relative activity (> 65% of maximum) at pH values ranging from 4.0 to 10.0 and the optimum pH was 8.0. The purified fibase exhibited maximal activity at 45 °C, and it was stable and retained over 90% of its initial activity after incubation for 1 h at temperatures between 28 °C and 50 °C. Even after incubation for 6 h at 50 °C the residual relative activity was > 75%.

Effects of pH and temperature on fibrinolytic enzyme activity and stability. All values are mean ± SD from three independent experiments
Effects of metal ions on fibase activity
The effects of different metal ions on fibase activity as determined via fibrin plates after dilution to various different concentrations are shown in Table 4. With the exception of activation by Ca2+ and Mg2+, all other metal ions investigated inhibited fibase activity.
Kinetic studies
The kinetic parameters Km and Vmax for fibase calculated using chromogenix substrate s-2251 as a substrate were 2.7 mmol/l and 0.03 mmol/l/min respectively (Fig. S3). The lower Km value of the purified fibase in the present study indicated that it had a greater affinity for the substrate.
Mechanisms of fibrinolytic effects and in vitro hemolysis assays
As shown in Fig. 7a and b the sizes of the translucent zones generated by purified enzyme on the two plates were very similar, but urokinase did not generate a translucent zone on the heated fibrin plate, suggesting that the mechanism of action of purified enzyme involves direct thrombolysis. Purified fibase did not form a translucent zone on the bovine blood agar plate, unlike the crude enzyme, indicating that the purified fibase did not induce hemolysis in vitro (Fig. 7c). Therefore, it may be a potential thrombolytic agent with low bleeding risk for safe therapy.

Mechanisms of fibrinolytic effects (a, b), and the hemolysis assay in vitro (c). In contrast to the urokinase (2), the purified enzyme (1) generated translucent zones of equal size on fibrin plates containing both plasminogen and fibrinogen (a) and fibrinogen only (b). The purified enzyme did not generate a translucent zone on a blood agar thus it did not induce hemolysis in vitro, unlike the crude extract (3)
Discussion
Codon optimization is a key efficient strategy and measure to improve the expression, which has been repeatedly demonstrated to increase heterologous protein expression in K. phaffii. The expressions of a codon-optimized lip2 gene from spergillus niger was enhanced 11.6-fold compared to the native gene [30]. Codon optimization of the endoinulinase gene resulted in 4.8-fold increases in enzyme activity [31]. Compared with the expression level of native hBMP4 sequence (12 mg/L), there is a 4-fold increase in that from the optimized sequence (48 mg/L) [32]. There are several factors remarkably affecting the expression of foreign genes in K. phaffii, containing codon usage bias, GC content, transcriptional and translational blocks, and secondary structure of mRNA et al. The optimization of these factors could finally result in an increase of protein synthesis.
The fib gene used in the current study was originated from a marine B. subtilis isolated from a coastal mangrove area in Beihai City in Guangxi, China. Marine microorganisms can survive in the extreme ocean environment, wherein the pH is similar to that of the internal environment of the human body. Therefore, marine microbes are considered a rich source of bioactive compounds that have high medicinal value [33, 34]. Since the 1960s thousands of novel secondary metabolites with antitumor, antiviral, antibacterial, antifungal, and anticoagulant activities have been isolated from marine microorganisms [35]. The recombinant fibase remained active over a broad pH (6.0–10.0) with optimum pH 8.0, which is very close to the physiological pH. Its optimum pH is quite different from many other fibrinolytic enzymes, but similar to fibrinolytic enzymes derived from high-salt environments, such as seas [36] and douchi [37]. Additionally, the purified fibase had high tolerance to the increasing temperature. As a metal-dependent serine protease, fibase did not cause hemolysis in vitro and preferentially degraded fibrin directly. All these characteristics exhibited its desirable adaptation to the internal environment of human body and great potential for application in thrombosis prevention and/or therapy.
An additional yield-limiting factor of recombinant protein is the efficiency of purification and recovery. Conventionally, separation and purification of wild fibase relies on methods like phenylene anion exchange, gel filtration, and hydrophobic interaction chromatography et al., which are complex operations resulting in low yields. Here, the fibase was purified by one-step Ni-NTA, achieving a 27 ± 2% recovery. The recovery was much higher than those of other fibrinolytic enzymes ever reported, such as 7.5% recovery from B. subtilis ICTF-1 [38], 6.46% recovery from Chive (Allium tuberosum) [6], and 7.82% recovery from Pseudomonas baetica SUHU25. This method is highly efficient and suited for large-scale purification of target protein.
Heterologous gene copy number reportedly plays an important role in the yield of recombinant proteins in K. phaffii expression systems [39]. In recent studies the activity of recombinant protein increased as the copy number increased from 4 to 9, but it decreased dramatically when the copy number continued to increase. That is a result of the negative effects of a high concentration of target protein on the growth and metabolism of the strain [40,41,42]. Song et al. (2019) reported that a co-expressing strain (pro/rDNA-mtg) with three copies of mtg genes (mtg-3c) exhibited higher transglutaminase activity than mtg-2c, mtg-6c, or mtg-8c [25]. Dagar et al. (2018) reported that the expression of human interleukin-3 protein increased with the addition of up to 8 copies of the expression cassettes, then drastically decreased thereafter [26]. Fang et al. (2014) reported that the respective maximum lipase activities of three recombinant strains with copy numbers of 7, 5, and 3 were 6600 U/ml, 6000 U/ml, and 4800 U/ml [27]. Therefore, the optimal copy number for high expression by K. phaffii needs to be evaluated via screening experiments, and it is not simply a case of “the more copies the better” [42]. After the optimal gene copy number has been evaluated, the main limitations of heterologous protein secretory expression pertain to protein translation, post-translational modification, and targeted delivery of protein precursors, which may be the main bottlenecks affecting yields [43, 44].
In order to provide a reference for the large-scale industrial production, the fermentation scale needs to be verified. There are many factors that affect fibrinolytic activity; thus the fermentation process needs prior optimization, which were initially performed at shake flask level to reduce cost and improve efficiency followed by fermenter study. As a complex and nutrient-rich medium, YPD free of glucose is prone to foaming. However, excessive antifoam may be toxic to cell growth, and to avoid this problem, an increase in inoculum size (10%) is required to allow cells to enter the log phase after 3 h. The rapid proliferation of K. phaffii resulted in faster consumption of available nutrients, and the decrease of medium foaming ability. Notably, K. phaffii showed transient secondary growth at 3 days, the reason of which may be that at least one nutrient had been used up and other nutrients started to be consumed. In addition, the initial induction biomass has been reported an important impact on recombinant protein yields [45]. If the cell density is too high, cell growth and metabolism will be restricted, resulting in a low enzyme activity; while if the cell density is too low, the induction phase will be much longer. The fibase activity of the strain expressing fib-9c was 12,690 U/ml after 3 days in a 5-L fermenter, which was 1.6-fold the highest fibase activity in shake flasks. The clear difference in fibase yield might attribute to the great difference between the fermentation conditions of fermenter and shaking flasks.
With the development of microbial technology, efficient protein expression could be achieved via genetic or engineering technologies. Some recently reported studies have investigated the homologous and heterologous expression of fibase, but their expression levels are typically low (Table 5). A study by Lv et al. overexpressed the recombinant plasmid pET-DsbA/PPFE-I in E. coli BL21 (DE3) and achieved maximum fibase activity of 228 U/mL after IPTG induction at 30 °C for 1.5 h, a level almost 2 times higher than that of wild strain P. polymyxa EJS-3 [8]. Fib gene (subtilisin DFE) fused with α-amylase promoter and signal peptide was successfully expressed in B. subtilis WB600, achieving a fibrinolytic activity of 200 U/mL. And it was 4-fold higher than that under the control of subtilisin DFE promoter [46]. Yao et al. transformed the fibase recombinant plasmid pHY300PLK/aprEBS15 into B. subtilis WB600 and achieved recombinant protein maximum fibrinolytic activity of 408.2 U/ml after induction at 37 °C for 96 h [47]. Several possible reasons might account for such a low production. First, the yield of recombinant fibase depends greatly on vector-host system. More specifically, high expression is generally associated with high-copy plasmids, and suitable host with high promoter activity plus high secretion efficiency. Second, the recombinant fibase might misfold into non-functional conformation, leading to aggregation or proteolytic digestion of misfolded protein. Third, the recombinant fibase may be toxic to the hosts, causing the host’s slow growth, impaired metabolism, or even death during expression. In order to improve the production of fibase in a high-efficiency K. phaffii expression system in the present study, various genetic and process engineering cultivation strategies were utilized at different levels. This resulted in improved fibase activity levels of up to 7930 U/ml in shake flasks and 12,960 U/ml in 5-L fermenter, which is among the highest levels of fibase production ever reported.
Conclusions
To conclude, in this work, the optimized fib gene was expressed at different copy numbers in K. phaffii to study the association between fibase production and gene dosage. Relative gene copy numbers were assessed by qPCR. The results showed that, the fibase levels increased as the copy number increased from 4 to 9, but decreased dramatically at copy number 12. The fermentation optimization of the strain expressing fib-9c was initially done at shake flask.
level and then fermenter verification, with the maximum fibase activity 7930 and 12,690 U/ml, respectively. The strategies developed in this work could be used to improve the expression of other therapeutic proteins that are difficult to express. The superior enzymatic properies of the purified fibase suggest that it has potential applications for the therapy and/or prevention of thrombosis.
Availability of data and materials
All the data presented in the article are available from the corresponding author upon reasonable request.
Abbreviations
- Fibase:
-
Fibrinolytic enzyme
References
Choi JH, Sapkota K, Park SE, Kim S, Kim SJ. Thrombolytic, anticoagulant and antiplatelet activities of codiase, a bi-functional fibrinolytic enzyme from Codium fragile. Biochimie. 2013;95:1266–77.
Blann AD, Landray MJ, Lip GYH. An overview of antithrombotic therapy. Brit Med J. 2002;325:762-5.
Wisner A. A novel plasminogen activator from Snake venom. J Biol Chem. 1995;270:10246–55.
Wu JX, Zhao XY, Pan R, He RQ. Glycosylated trypsin-like proteases from earthworm Eisenia fetida. Int J Biol Macromol. 2007;40:399–406.
Iannucci NB, Camperi SA, Cascone O. Purification of lumbrokinase from Eisenia fetida using aqueous two-phase systems and anion-exchange chromatography. Sep Purif Technol. 2008;64:131–4.
Chung DM, Choi NS, Maeng PJ, Chun HK, Kim SH. Purification and characterization of a novel fibrinolytic enzyme from chive (Allium tuberosum). Food Sci Biotechnol. 2010;19:697–702.
Katrolia P, Liu X, Zhao Y, Kopparapu NK. Zheng. Gene cloning, expression and homology modeling of first fibrinolytic enzyme from mushroom (Cordyceps militaris). Int J Biol Macromol. 2019;146:897–906.
Lv FX, Zhang C, Guo FF, Lu YJ, Bie XM, Qian H, Lu ZX. Expression, purification, and characterization of a recombined fibrinolytic enzyme from endophytic Paenibacillus polymyxa EJS-3 in Escherichia coli. Food Sci Biotechnol. 2015;24:125–31.
Taneja K, Bajaj BK, Kumar S, Dilbaghi N. Production, purification and characterization of fibrinolytic enzyme from Serratia sp. KG-2-1 using optimized media. 3 Biotech. 2017;7:1–15.
Wong SL. Advances in the use of Bacillus subtilis for the expression and secretion of heterologous proteins. Cur Opin Biotech. 1995;6:517–22.
Lam KHE, Chow KC, Wong WKR. Construction of an efficient Bacillus subtilis system for extracellular production of heterologous proteins. J Biotechnol. 1998;63:167–77.
Westers L, Dijkstra DS, Westers H, van Dijl JM, Quax WJ. Secretion of functional human interleukin-3 from Bacillus subtilis. J Biotechnol. 2006;123:211–24.
Kodama T, Manabe K, Kageyama Y, Liu S, Ara K, Ozaki K, Sekiguchi J. Approaches for improving protein production in multiple protease-deficient Bacillus subtilis host strains. Adv Appl Microbiol. 2012;8:163-76.
Celik E, Calik P. Production of recombinant proteins by yeast cells. Biotechnol Adv. 2012;30:1108–18.
Idiris A, Tohda H, Kumagai H, Takegawa K. Engineering of protein secretion in yeast: strategies and impact on protein production. Appl Microbiol Blot. 2010;86:403–17.
Liu YK, Huang H. Expression of single-domain antibody in different systems. Appl Microbiol Biot. 2018;102:539–51.
Eissazadeh S, Moeini H, Dezfouli MG, Heidary S, Nelofer R, Abdullah MP. Production of recombinant human epidermal growth factor in Pichia pastoris. Braz J Microbiol. 2017;48:286–93.
Barathiraja S, Gangadhara PAV, Umapathi V, Dechamma HJ, Reddy GR. Expression and purification of biologically active bovine interferon lambda3 (IL28B) in Pichia pastoris. Protein Express Purif. 2018;145:14–8.
Yu KM, Lau JYN, Fok M, Yeung YK, Fok SP, Shek F, Wong WT, Choo QL. Efficient expression and isolation of recombinant human interleukin-11 (rhIL-11) in Pichia pastoris. Protein Express Purif. 2018;146:69–77.
Polez S, Origi D, Zahariev S, Guarnaccia C, Tisminetzky SG, Skoko N, Baralle M. A simplified and efficient process for insulin production in Pichia pastoris. PLoS One. 2016;11:1–15.
Vandana Kantipudi S, Maheshwari N, Sharma S, Sahni G. Cloning and purification of an anti-thrombotic, chimeric Staphylokinase in Pichia pastoris. Protein Expres Purif. 2019;162:1–8.
Yang Z, Zhang Z. Engineering strategies for enhanced production of protein and bio-products in Pichia pastoris: a review. Biotechnol Adv. 2018;36:182–95.
Daly R, Hearn MT. Expression of heterologous proteins in Pichia pastoris: a useful experimental tool in protein engineering and production. J Mol Recognit. 2005;18:119–38.
Macauley-Patrick S, Fazenda ML, McNeil B, Harvey LM. Heterologous protein production using the Pichia pastoris expression system. Yeast. 2005;22:249–70.
Song XP, Shao CS, Guo YG, Wang YJ, Cai JJ. Improved the expression level of active transglutaminase by directional increasing copy of mtg gene in Pichia pastoris. BMC Biotechnol. 2019;19:1–9.
Dagar VK, Khasa YP. Combined effect of gene dosage and process optimization strategies on high-level production of recombinant human interleukin-3 (hIL-3) in Pichia pastoris fed-batch culture. Int J Biol Macromol. 2018;108:999–1009.
Fang ZG, Xu L, Pan DJ, Jiao LC, Liu ZY, Yan YJ. Enhanced production of Thermomyces lanuginosus lipase in Pichia pastoris via genetic and fermentation strategies. J Ind Microbiol Blot. 2014;41:1541–51.
Waterham HR, Digan ME, Koutz PJ, Lair SV, Cregg JM. Isolation of the Pichia pastoris glyceraldehyde-3-phosphate dehydrogenase gene and regulation and use of its promoter. Gene. 1997;186:37–44.
Astrup T, Müllertz S. The fibrin plate method for estimating Fibrinolytic activity. Arch Biochem Biophys. 1952;40:346–51.
Yang J, Liu LY. Codon optimization through a two-step gene synthesis leads to a high-level expression of Aspergillus Niger lip2 gene in Pichia pastoris. J Mol Catal B Enzym. 2010;63:164–9.
He M, Wu D, Wu J, Chen J. Enhanced expression of endoinulinase from Aspergillus niger by codon optimization in Pichia pastoris and its application in inulooligosaccharide production. J Ind Microbiol Biot. 2014;41:105–14.
Huang Y, Zhen BQ, Lin Y, Cai YH, Lin Z, Deng CM, Zhang YD. Expression of codon optimized human bone morphogenetic protein 4 in Pichia pastoris. Biotechnol Appl Bioc. 2014;61:175–83.
Ngo DH, Vo TS, Ngo DN, Wijesekara I, Kim SK. Biological activities and potential health benefits of bioactive peptides derived from marine organisms. Int J Bilo Macromol. 2012;51:378–83.
Romano G, Costantini M, Sansone C, Lauritano C, Ruocco N, Ianora A. Marine microorganisms as a promising and sustainable source of bioactive molecules. Mar Environ Res. 2017;128:58–69.
Carroll AR, Copp BR, Davis RA, Keyzers RA, Prinsep MR. Marine natural products. Nat Prod Rep. 2019;36:122–73.
Vijayaraghavan P, Prakash Vincent SG. A low cost fermentation medium for potential fibrinolytic enzyme production by a newly isolated marine bacterium, Shewanella sp. IND20. Biotechnol Rep. 2015;7:135–42.
Hu YL, Yu D, Wang ZT, Hou JJ, Tyagi R, Liang YX, Hu YM. Purification and characterization of a novel, highly potent fibrinolytic enzyme from Bacillus subtilis DC27 screened from Douchi, a traditional Chinese fermented soybean food. Sci Rep-UK. 2019;9:1–10.
Mahajan PM, Nayak S, Lele SS. Fibrinolytic enzyme from newly isolated marine bacterium Bacillus subtilis ICTF-1: media optimization, purification and characterization. J Biosci Bioeng. 2012;113:307–14.
Zhu T, Guo M, Tang Z, Zhang M, Zhuang Y, Chu J, Zhang S. Efficient generation of multi-copy strains for optimizing secretory expression of porcine insulin precursor in yeast Pichia pastoris. J Appl Microbiol. 2009;107:954–63.
Marx H, Mecklenbrauker A, Gasser B, Sauer M, Mattanovich D. Directed gene copy number amplification in Pichia pastoris by vector integration into the ribosomal DNA locus. FEMS Yeast Res. 2009;9:1260–70.
Javitt G, Ben-Barak-Zelas Z, Jerabek-Willemsen M, Fishman A. Constitutive expression of active microbial transglutaminase in Escherichia coli and comparative characterization to a known variant. BMC Biotechnol. 2017;17:1–10.
Juturu V, Wu JC. Heterologous protein expression in Pichia pastoris. Latest Research Progress and Applications Chembiochem. 2018;19:7–21.
Li F, Yu XW, Sha C, Xu Y. Impact of gene dosage and methanol concentration on Rhizopus chinensis recombinant lipase production in Pichia pastoris. Microbiology China. 2011;38:301–9.
Li A, Ilie A, Sun Z, Lonsdale R, Xu JH, Reetz MT. Whole-cell-catalyzed multiple Regio- and Stereoselective Functionalizations in Cascade reactions enabled by directed evolution. Angew Chem. 2016;55:12026–9.
Liu WC, Gong T, Wang QH, Liang X, Chen JJ, Zhu P. Scaling-up fermentation of Pichia pastoris to demonstration-scale using new methanol-feeding strategy and increased air pressure instead of pure oxygen supplement. Sci Rep-UK. 2016;6:1–12.
Xiao L, Zhang RH, Peng Y, Zhang YZ. Highly efficient gene expression of a fibrinolytic enzyme (subtilisin DFE) in Bacillus subtilis mediated by the promoter of α-amylase gene from Bacillus amyloliquefaciens. Biotechnol Lett. 2004;26:1365–9.
Yao Z, Kim JA, Kim JH. Gene cloning, expression, and properties of a Fibrinolytic enzyme secreted by Bacillus pumilus BS15 isolated from Gul (oyster) Jeotgal. Biotechnol Bioproc E. 2018;23:293–301.
Vijayaraghavan P, Arasu MV, Anantha Rajan R, Al-Dhabi NA. Enhanced production of fibrinolytic enzyme by a new Xanthomonas oryzae IND3 using low-cost culture medium by response surface methodology. Saudi J Biol Sci. 2019;26:217–24.
Anusree M, Swapna K, Aguilar CN, Sabu A. Optimization of process parameters for the enhanced production of fibrinolytic enzyme by a newly isolated marine bacterium. Bioresour Technol Rep. 2020;11:1–7.
Vijayaraghavan P, Rajendran P, Vincent SGP, Arun A, Al-Dhabi NA, Arasu MV, Kwon OY, Kim YO. Novel Sequential Screening and Enhanced Production of Fibrinolytic Enzyme by Bacillus sp IND12 Using Response Surface Methodology in Solid-State Fermentation. Biomed Res Int. 2017;2017:1–13.
Liu X, Kopparapu NK, Li Y, Deng Y, Zheng X. Biochemical characterization of a novel fibrinolytic enzyme from Cordyceps militaris. Int J Biol Macromol. 2017;94(Pt B):793–801.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (31560448, 21062001) and the Natural Science Foundation of Guangxi (2016GXNSFAA380130).
Author information
Authors and Affiliations
Contributions
ZL and ZC contributed in the conception and design of the research; ZC carried out the experiments and wrote the manuscript; XC and GC contributed to analysis, or interpretation of data; ZL revised the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
Sequence of optimized and original fib gene. Upper row: the optimized fib sequence, lower row: the original fib sequence, different nucleic acids are marked with red.
Additional file 2: Fig. S2.
Detection of fib copy number in the K. phaffii genome via a double standard curve method. (A) and (B) are the melting curves of TDH1 and fib genes, and (C) and (D) are the amplification curves of TDH1 and fib genes.
Additional file 3: Fig. S3.
Lineweaver–Burk plots for purified fibrinolytic enzyme using chromogenix as a substrate. The experiment was repeated three times independently.
Additional file 4: Table S1.
Important parameters during the entire fermentation process.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Che, Z., Cao, X., Chen, G. et al. An effective combination of codon optimization, gene dosage, and process optimization for high-level production of fibrinolytic enzyme in Komagataella phaffii (Pichia pastoris). BMC Biotechnol 20, 63 (2020). https://doi.org/10.1186/s12896-020-00654-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12896-020-00654-7