
Abstract
Background
Whey permeate is a lactose-rich effluent remaining after protein extraction from milk-resulting cheese whey, an abundant dairy waste. The lactose to ethanol fermentation can complete whey valorization chain by decreasing dairy waste polluting potential, due to its nutritional load, and producing a biofuel from renewable source at the same time. Wild type and engineered microorganisms have been proposed as fermentation biocatalysts. However, they present different drawbacks (e.g., nutritional supplements requirement, high transcriptional demand of recombinant genes, precise oxygen level, and substrate inhibition) which limit the industrial attractiveness of such conversion process. In this work, we aim to engineer a new bacterial biocatalyst, specific for dairy waste fermentation.
Results
We metabolically engineered eight Escherichia coli strains via a new expression plasmid with the pyruvate-to-ethanol conversion genes, and we carried out the selection of the best strain among the candidates, in terms of growth in permeate, lactose consumption and ethanol formation. We finally showed that the selected engineered microbe (W strain) is able to efficiently ferment permeate and concentrated permeate, without nutritional supplements, in pH-controlled bioreactor. In the conditions tested in this work, the selected biocatalyst could complete the fermentation of permeate and concentrated permeate in about 50 and 85 h on average, producing up to 17 and 40 g/l of ethanol, respectively.
Conclusions
To our knowledge, this is the first report showing efficient ethanol production from the lactose contained in whey permeate with engineered E. coli. The selected strain is amenable to further metabolic optimization and represents an advance towards efficient biofuel production from industrial waste stream.
Similar content being viewed by others
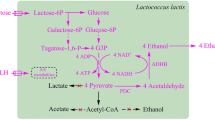

Background
Whey is an abundant waste stream generated during cheese production. After cheese curdling, about 10% of the used milk is converted in cheese, while the remaining liquid is a by-product called whey, which still contains about 55% of the milk nutritional load [1, 2]. In particular, although whey composition depends on several factors (e.g., milk quality, animal breed and feed), a high lactose concentration (about 45 g/l), and about 6–10 g/l of proteins are usually present [3,4,5,6]. They correspond to the total amount of milk lactose content and about 20% of milk proteins, respectively [2].
Worldwide, 160 million tons of whey per year are produced [2]. It represents an environmental problem for its high nutritional load, largely due to lactose content, and, for this reason, it cannot be discharged in water systems without pre-treatments [3]. Several options, extensively reviewed by Prazeres et al. [4], are available to decrease the organic content of whey and, in some cases, valorize this waste to obtain added-value bioproducts at the same time: biological treatments with or without valorization, physicochemical treatment and direct land application. Valorization can be carried out by recovering high-value biomolecules, such as the protein fraction that is separated via ultrafiltration or diafiltration, obtaining whey protein concentrates which can be used in food, cosmetic and pharmaceutical industries. The liquid remaining after this process (called whey permeate - WP) has the same lactose concentration as whey and for this reason its pollution load is still high [4]. Furthermore, WP can be concentrated to facilitate its transportation to treatment plants, obtaining concentrated whey permeate (CWP) that can have a lactose concentration up to about 160 g/l. Extraction of lactose from whey or WP is not always economically convenient. Hence, lactose to ethanol fermentation is considered as a further treatment of WP, required to decrease its pollutant load, simultaneously enabling the production of a commercially attractive biomolecule from waste material [2, 4, 7]. Ethanol can be used as a fuel, but also in food and beverages, pharmaceutical and cosmetic industries.
In large-scale production plants, ethanol is normally obtained from sugars (e.g. sucrose molasses and hydrolyzed starch) by alcoholic fermentation of the baker’s yeast Saccharomyces cerevisiae or the soil bacterium Zymomonas mobilis, which are not able to ferment lactose [2, 8]. Enzymatic or chemical pre-hydrolysis of lactose into glucose and galactose and subsequent feeding of S. cerevisiae with these sugars can be carried out [9], but it is not economically convenient for the high cost of the required enzymes or chemical pre-process [2, 10] and for the catabolite repression phenomenon [11]. Strains affected by such phenomenon show slower fermentations of sugar mixtures, such as glucose and galactose, compared to strains without catabolite repression, although mutant yeasts not exhibiting this phenotype can be selected [12]. Naturally occurring organisms able to ferment lactose into ethanol include Torula cremoris, Kluyveromyces fragilis, Kluyveromyces lactis, Kluyveromyces marxianus and Candida pseudotropicalis yeasts [2]. However, despite examples of industrial implementation of whey-to-ethanol production plants using whey or deproteinated whey as substrate are present [2], such microorganisms are not ideal workhorses for the ethanol production industry because several drawbacks affect their use in large-scale plants, such as low productivity, metabolic inhibition at high lactose concentration, complex nutritional requirements and impaired growth in low-oxygen conditions [2]. Because of such drawbacks, research is still ongoing to optimize the underlying conversion processes and test different naturally-occurring strains as biocatalysts [2, 5, 13, 14]. For the above reasons, whey-to-ethanol fermentation technology requires a dramatic improvement in order to enhance the attractiveness of the bioprocess [2, 7].
Engineered microorganisms able to convert lactose to ethanol have been constructed. The main examples include metabolic modifications of ethanol-producing baker’s yeast to enable lactose fermentation, and of lactose-consuming bacteria to enable ethanol production from pyruvate. Since this work focuses on the latter, particularly on Escherichia coli, the main literature outcomes are briefly reported, while the efforts to create metabolically engineered S. cerevisiae are reviewed elsewhere [2]. Wild type E. coli is able to ferment lactose to produce a mix of organic acids and a low amount of ethanol [15]. Efforts have been carried out in the last 30 years to build recombinant E. coli expressing the Z. mobilis pdc and adhB genes (encoding pyruvate decarboxylase and alcohol dehydrogenase, respectively) to direct pyruvate metabolism towards ethanol production, by the pyruvate to acetaldehyde and carbon dioxide reaction via pdc, and the acetaldehyde to ethanol reaction via adhB. The two ethanologenic genes have been assembled with their native ribosome binding sites (RBSs) in an operon and expressed under the control of different promoters via multicopy plasmids [16,17,18] or integrated in the genome [19] of strains previously tested for their environmental hardiness and substrate fermentation range [18], with the final aim of constructing a biocatalyst for efficient fermentation of plant biomass sugars [20,21,22]. The strains constructed in literature often showed high transcriptional demand for pdc and adhB, which needed very strong promoters, multicopy plasmids or multiple tandem chromosomal repeats to be properly expressed [19, 23,24,25]. For instance, KO11, the first constructed and most widely used E. coli strain with pdc and adhB in the chromosome [19], has about 25 copies of the ethanologenic genes [26], generated via antibiotic selection procedure to obtain high ethanol production, which was low in the initial clone. The transcriptional demand may also affect the genetic stability of the genes [15, 20, 27, 28], and represents a problem when dealing with scenarios typical of real industrial settings, like the use of poor media, in which nutrients can be insufficient for suitable expression of ethanologenic genes [23, 29]. Expensive nutrient supplementations could be avoided in minimal media by using other strains that have been proposed by re-engineering existing biocatalysts using random integration sites and selection based on growth and ethanol production [30] or re-engineering different hosts [24]. Strains were also engineered for algal biomass fermentation [31]. Despite no E. coli strain was specifically engineered to ferment dairy waste, examples of successful ethanol production from cheese whey via the KO11 strain have been reported and nutrient supplementation was needed to obtain a reasonable fermentation performance [32, 33]. Finally, engineered strains have also been constructed, again mainly focused on cellulosic biomass fermentation, using different strategies, such as laboratory metabolic evolution [34], rational pathway engineering supported by elementary mode analysis [35], stabilization of plasmid-borne pdc-adhB via mutations that can be complemented only via ethanologenic genes in strict anaerobic growth [36, 37], and genome engineering without foreign genes [38, 39].
In summary, a number of studies have reported the successful construction and characterization of ethanologenic E. coli strains, but no specific study has been carried out to construct an ad-hoc strain for dairy waste fermentation. The studies above shed light on many critical issues that can support the engineering of future ethanologenic microbes. In this work, we aim to metabolically engineer a set of candidate Escherichia coli strains (reported in Table 1) via a new pdc-adhB expression plasmid, and we carry out the selection of the best strain in terms of growth in permeate, lactose consumption and ethanol formation. We finally show that the selected engineered microbe is able to efficiently ferment WP and CWP, without nutritional supplements, in pH-controlled bioreactor.
It is worth noting that the search of optimal fermentation parameters is not addressed in this work. An exploratory analysis is herein carried out by testing a number of experimental conditions, which represent only a subset of all the possible ones, to test WP and CWP fermentation feasibility. A systematic parameter search for the selected strain will be investigated in future studies.
Results
pL13 construction and preliminary characterization in LB
The pL13 expression plasmid was designed and constructed to meet the following specifications: an operon structure with adhB-pdc driven by the regulated Plux promoter (BBa_R0062 from the Registry of Standard Biological Parts) [40]; strong RBSs (BBa_B0030 from the Registry of Standard Biological Parts) [41] assembled upstream of both genes; a low-copy number replication origin, enabling plasmid maintenance at 3–7 copies per cell [42, 43]. Together with the codon-optimized design of pdc and adhB, the above specifications aim to maximize the translational efficiency of the two recombinant genes, to reduce the high transcriptional demand reported in the literature for previously developed strains. Although the quantitative levels of gene expression and protein synthesis are hard to predict in operon architecture and many context-dependent features can affect them [44], software tools are available to support their forward and reverse engineering. A recently proposed tool for sequence-to-function prediction, based on biophysical model of translational coupling (Operon Calculator [45]), estimated a 10- and 90-fold improvement in translation initiation rate of pdc and adhB, respectively, in our synthetic operon compared to the operon integrated in the KO11 genome [26]. Finally, in our operon, the Plux promoter also enables downstream mRNA expression tuning over a wide range of transcriptional activities to probe optimal enzyme levels if required [40]. In this work, only the Plux basic activity was exploited for pdc and adhB expression.
The pL13 plasmid was initially transformed in MG1655, a widely used K-12 strain, to perform preliminary experiments. Enzymatic assays for pdc and adhB showed a successful expression of both enzymes (Table 2). Fermentation experiments in LB supplemented with 40 g/l of lactose in 15-ml tubes demonstrated that the engineered MG1655 could produce high levels of ethanol (17 g/l) in 72 h at 30 °C, consuming more than half of the available lactose (Fig. 1a). The non-engineered strain, conversely, produced less than 1 g/l of ethanol and consumed a much lower amount of lactose. The engineered MG1655 also showed typical traits of ethanologenic strains [16]: higher pH (6 versus 5, despite the presence of phosphate buffer) and cell concentration (OD600 values of ~1.0 versus ~0.5, see Methods section for details about cell density measurements) at the end of fermentation, compared to the non-engineered strain. The higher pH is the result of the decrease of organic acid production fluxes from pyruvate, caused by the recombinant pathway introducing a pyruvate to ethanol route. At the reached concentrations, ethanol is less toxic than the organic acids produced in the wild type fermentation pathway [17] and the recombinant cultures could reach a higher cell density than the non-engineered MG1655. Fermentation experiments in LB + 80 g/l of lactose confirmed the ethanol production performance (Fig. 1b), pH (6 versus 5) and growth (~1.0 versus ~0.6) of MG1655 with pL13. Although these preliminary experiments demonstrated the successful functioning of the recombinant pathway, they highlighted an incomplete lactose consumption, probably due to the lack of a strict pH control, which can inhibit cell growth and ethanol production [46].

Preliminary fermentation experiments with wild type and engineered MG1655 in LB with phosphate buffer at the indicated lactose concentration in 15-ml tubes. a 40 g/l of lactose. b 80 g/l of lactose. Ethanol and residual lactose are measured after a 72-h fermentation at 30 °C. The reported values are the mean of at least 4 independent replicates and error bars are the 95% confidence intervals of the mean
Strain selection
After MG1655 engineering, the pL13 plasmid was incorporated into other seven E. coli strains (Table 1), which were then tested in WP in terms of growth, lactose consumption and ethanol formation to select the best biocatalyst for fermentation. The eight strains of Table 1 were selected to meet these main specifications for an efficient biocatalyst: non-pathogenic strains able to consume lactose, possibly used previously for ethanol production from different substrates (meaning that the strain is amenable for genetic modifications and may be also suitable for ethanol production from dairy waste) [18, 19, 24, 30, 31, 35, 38, 39] and possibly wild type strains (suggesting that they can display a fast-growth phenotype without auxotrophies, compared with highly engineered laboratory strains). All the regulatory parts of the pL13 expression plasmid, i.e., promoter, RBSs and low-copy replication origin, were tested as previously reported [47, 48] and they resulted to be fully functional in the eight candidate strains (data not shown).
Growth assays in 96-well microplates demonstrated that all the strains could successfully grow in WP (Fig. 2a-b). Doubling times in exponential growth phase exhibited a narrow range (0.4–0.75 h, considering average data), and did not show relevant differences between engineered and non-engineered strains (Fig. 2c). The maximum cell density reached in a 21-h growth time showed that engineered strains could reach higher cell densities (1.4-fold on average) than the wild types, as expected (Fig. 2d). Moreover, maximum cell density spanned a 5-fold range (0.11–0.49, considering average data) among different strain backgrounds, with the W strain showing the highest OD600, followed by the Crooks and W3110 strains. The B strain showed the lowest OD600.

Growth curves in WP for the eight candidate strains in 96-well microplates experiments. a Growth assay results for the strains engineered with pL13. b Growth assay results for the non-engineered strains. c Doubling time in exponential growth phase. d Maximum OD600 reached in the experiment. Data points in panels a-b and bars in panels c-d represent the average values of at least two independent replicates. Error bars in panels c-d represent the 95% confidence intervals of the mean (due to the presence of an outlier, exhibiting noisy measurements in the exponential growth phase, no replicates were available to compute confidence intervals for the wild-type Crooks strain in panel c)
Fermentation experiments in 15-ml tubes, carried out in WP at two temperatures (30 °C, measured at 72 h and 168 h, and 37 °C, measured at 72 h, see Fig. 3), showed that all the engineered strains could successfully produce ethanol from lactose. The ML308 strain, followed by the W strain, gave the best performance, while the C and Crooks strains had the lowest one. In particular, 15.2 g/l of ethanol were produced by the ML308 strain in 168 h, with a residual lactose of less than 1 g/l, while 13.8 g/l of ethanol were produced by the W strain in the same conditions, with less than 8 g/l of residual lactose. Data after 72 h also confirmed that ML308 and W were superior, with about 9 g/l of ethanol and 18 g/l of residual lactose for both strains. Enzymatic activities were also measured for these two strains (Table 2). As expected, a correlation was present between pdc and adhB activities in each strain with pL13 (MG1655, W and ML308), since the two recombinant genes are assembled in the same operon. A >5-fold activity range was observed in the three different strain backgrounds for both enzymes (0.156–1 for pdc and 0.17–1 for adhB), confirming that they were properly expressed and also supporting previous findings that the same expression plasmid for pdc and adhB could give highly different enzyme activities in different hosts [18].

Fermentation results in WP for the eight candidate strains with pL13 in 15-ml tubes. a Ethanol production. b Residual lactose. Bars represent the average values of 2 to 4 independent experiments. Error bars represent the 95% confidence intervals of the mean. The initial lactose concentration of the used WP batch was about 45 g/l
Considering all strains after 72 h, the mean ethanol concentration was slightly higher at 37 °C than at 30 °C (9.1 versus 7.6 g/l; p-value < 0.05, paired t-test).
The maximum ethanol concentration reached by the Crooks and C strains in the best condition (37 °C at 72 h) was 2-fold lower than the one reached by the two strains exhibiting superior performance (W and ML308), and a considerably high residual lactose was also left (>25 g/l). Among the other strains, it is worth noting that the B strain also showed high ethanol production (13.5 g/l) and low residual lactose (8.4 g/l) after 168 h, but its performance after 72 h was poor, probably for its poor growth capability in WP (see Fig. 2a). This strain was already used as a host for ethanol production with promising results [18, 39], also from lactose [18], but the results shown here highlight the need for a specific screening for ethanol production and sugar consumption in dairy waste. The Crooks strain was also used in literature as an efficient ethanol producer from different sugars (not lactose), but we could not observe reasonably good performance in the conditions tested here (see Fig. 3), despite its promising results in terms of growth capability (see Fig. 2d).
Considering the growth and fermentation results, the W and ML308 strains were selected for further study.
Fermentation of permeate and concentrated permeate in pH-controlled bioreactor
The parallelized assays carried out so far did not enable the characterization of fermentation performance in conditions compatible with industrial settings, in which pH control is essential to maximize bioconversion efficiency parameters, such as maximum product concentration, fermentation yield and volumetric productivity [46]. In a preliminary pH-controlled bioreactor experiment for the W strain carried out in LB + 80 g/l of lactose at 30 °C, pH 7.0, we observed a high ethanol production (29 g/l) after only 1 day, with a complete lactose consumption (Additional file 1: Figure S1), corresponding to 70% of the maximum theoretical yield (the main fermentation parameters, including the ones mentioned above, are shown in Table 3 for each experiment of this study). We then carried out pH-controlled exploratory experiments in WP to evaluate the fermentation feasibility and performance in different pH, temperature and engineered strain contexts (Fig. 4). In the tested conditions, we found that the engineered W strain could consume all the lactose and convert it into ethanol with a 54–65% conversion yield. The ML308 strain, tested at 30 °C, pH 7.0, showed slightly lower conversion yield and productivity than the W strain in the same conditions (51% versus 60% and 0.13 versus 0.15 g/l/h). According to the data in Fig. 4 and Table 3, we selected the 37 °C, pH 6.6 condition for further investigation, despite a slightly lower fermentation yield than in the test at 30 °C, already observed in other ethanologenic E. coli with different fermentation substrates [46]. In particular, the fast conversion obtained at 37 °C (also highlighted by the 2-fold increase in volumetric productivity from the test at 30 °C to the one at 37 °C, both at pH 7.0, see Table 3) is attractive in industrial context, since a decrease in process time can result in a more rapid turnover of the fermentation tanks, even if the increased energy demand and slight yield decrease should be considered. One of the conditions above (W strain, 37 °C, pH 7.0) was also scaled-up by 8-fold in a 2.4-liter volume without performing filter-sterilization of WP. Results showed analogous fermentation time, maximum ethanol concentration and productivity to the small-scale experiment (Fig. 4 and Additional file 1: Figure S2). Although the lactose to ethanol conversion yield in WP was lower than the one observed in a rich medium like LB (70%, see Additional file 1: Figure S1 and Table 3), these results are promising and a complete lactose consumption was always observed, producing 11.7 to 17.6 g/l of ethanol in the tested conditions.

Fermentation of WP in a pH-controlled bioreactor. Ethanol and lactose concentrations over time are shown for different conditions in terms of temperature (30 °C or 37 °C), pH (6.6 or 7.0) and host strain (W or ML308) bearing pL13
We finally carried out three fermentation experiments for the engineered W strain in CWP (Fig. 5a). The use of a waste with a higher lactose concentration than WP is industrially attractive from an economical point of view, since a higher amount of ethanol will be present in the fermentation broth and distillation costs per liter of ethanol will decrease. However, the possible additional costs of the concentration process should also be considered. Results show that the engineered W strain can also efficiently ferment CWP with a 45–64% conversion yield, producing up to 40 g/l of ethanol and consuming all the lactose (117 to 137 g/l according to the three waste batches) in about 70 (two replicates) to 116 (one replicate) h. These data demonstrate that, although conversion yield could be further improved and a relevant variability is displayed, the lactose to ethanol fermentation in WP and CWP is feasible via an ad-hoc selected engineered bacterial strain without nutritional supplements. The engineered ML308 strain was also tested in the same conditions and the results of two experiments (Fig. 5b) showed performance comparable with the engineered W strain in terms of conversion yield (39–62%) and maximum ethanol concentration (up to 36 g/l). Despite a relatively low conversion yield was observed in the first experiment with ML308 (39%), the measured volumetric productivity (0.72 g/l/h) was almost 2-fold higher than in the other replicate and in the experiments with the W strain. By comparing the average productivity and conversion yield between the two strains, no statistically significant difference was found (p-value > 0.05, t-test).

Fermentation of CWP in a pH-controlled bioreactor for W-pL13 (a) and ML308-pL13 (b). Ethanol and lactose concentrations over time are shown for three (a) or two (b) independent experiments carried out at 37 °C, pH 6.6
The W strain was also transformed with pLOI297 (ATCC 68239, prepared according to ATCC instructions), a high-copy number plasmid for the high-level expression of wild-type pdc-adhB, used for efficient ethanol production in different published works [18, 35, 46]. The resulting engineered strain (W-pLOI297) was tested in CWP. Results showed a comparable performance with our W-pL13 strain (Table 3 and Additional file 1: Figure S3), although the activity of pdc and adhB was much higher with pLOI297 than with pL13 (Table 2). Such data suggest that the basic activity of the Plux promoter in pL13 is sufficient to drive the expression of pdc and adhB at suitable levels for ethanol production in WP and CWP, and the use of a plasmid providing higher activity levels of both enzymes does not seem to result in a clear improvement of fermentation performance.
Discussion
In this work, a new engineered biocatalyst has been constructed, with design features specific for ethanol production from dairy waste. To our knowledge, this is the first report showing ethanol production from the WP and CWP lactose with engineered E. coli. The main steps of this work included the construction of a new expression plasmid, strain selection in parallelized experiments in WP and CWP, and pH-controlled fermentation tests. The new plasmid (pL13) includes a fully synthetic operon for pdc and adhB expression, engineered to maximize the translation of the two proteins. Translation was maximized to overcome the high transcriptional demand previously observed for wild-type ethanologenic genes that often needed to be expressed at high levels or placed at high DNA copy numbers, often via tandem chromosomal repeats amplification, to enable sufficient ethanol production [19, 23, 25, 26], and for these reasons also resulting in unstable systems [20, 27]. The maximization of translation was carried out via codon optimization and use of efficient RBSs. The basic activity of a regulated promoter, Plux, in the off-state was used to drive the expression of pdc and adhB in pL13. We planned to use such promoter to produce a versatile plasmid for pdc and adhB expression tuning via chemical inducer (N-3-oxohexanoyl-L-homoserine lactone), but we found that the basic transcriptional activity already gave reasonably good performance in the engineered W and ML308 strains. For this reason, no inducer was added in this work. The fermentation performance of W-pLOI297 was comparable to the one of our W-pL13 strain, supporting the assumption that the pdc and adhB levels produced by pL13 are sufficient for an efficient ethanol production in the considered dairy waste. Nonetheless, their activity is much lower in pL13 than in pLOI297 and previous works reported correlation between enzyme activity and fermentation yield [17, 23]. While the major aim of this work was to demonstrate the feasibility of WP and CWP fermentation via ad-hoc selected engineered E. coli, further work should be carried out on our new codon-optimized genes to understand their optimal expression level and try to further improve ethanol yield.
Strain selection process resulted in the choice of the W and ML308 strains as best engineered biocatalysts, according to their growth and fermentation performance. Although these two strains were already considered as efficient ethanol production systems, their choice among the candidate strains was not trivially predictable. In fact, other strains that have been extensively used in literature as efficient biocatalysts were excluded: the B strain showed poor growth in WP and high ethanol levels only after 168 h, while the Crooks strain showed high level growth but low ethanol production. These results further demonstrate the need of strain selection for the specific task and process to be carried out.
Tests in pH-controlled bioreactor were carried out considering both W and ML308 strains, which showed comparable performance in terms of fermentation profiles and parameters. However, we identified the engineered W strain as the best candidate between the two strains for two main reasons: i) it is more amenable to further metabolic engineering steps [49], since its genome is fully available [50]; ii) its transformation efficiency is much higher than ML308, which was the only strain among the ones in Table 1 that showed poor efficiency (data not shown), also by using a different transformation protocol [51].
The lactose to ethanol conversion efficiency did not considerably vary among the experiments carried out in dairy waste in different conditions. However, it is worth noting that several problems might affect fermentation performance in industrial context, such as waste composition and contaminations. In our experiments, initial concentration of lactose showed variability in both WP and CWP (see Table 3). Given a condition, fermentation efficiency itself can vary, as shown in the experiments with two or three replicates in CWP. Additional studies concerning fermentation (e.g., pH and temperature tuning) and bioprocess (e.g., inoculum size and preparation) optimization will be needed to demonstrate not only the feasibility of CWP fermentation via engineered E. coli, but also its economic competitiveness compared with other processes with different biocatalysts. One preliminary test was successfully carried out in this work using a 2.4-liter culture without sterilization of WP, thus providing promising results concerning contamination problems. When the optimal enzyme levels, fermentation parameters and bioprocess steps are defined, a proper number of experimental replicates and long-term operation times will provide useful insights into the real sustainability of the process. In this work, a number of conditions was tested, but a systematic search of optimal fermentation parameters was not addressed and additional investigations will be performed. Here, only an exploratory analysis was carried out, testing a subset of all the possible conditions, to support strain selection (in 15-ml tubes and 96-multiwell plates experiments) and to test the feasibility of WP and CWP fermentation (in pH-controlled experiments). In particular, strain growth was assayed in WP in 96-multiwell plates via OD600 measurements; ethanol and lactose concentrations were measured in 15-ml tubes fermentations at two temperatures after 72 h, and only one of the conditions was also tested after 168 h to evaluate if prolonged fermentation time could improve fermentation performance. Analogously, pH-controlled fermentations in WP were carried out in three temperature/pH conditions without testing a full factorial design, and the parameter values associated with improved fermentation performance were chosen for the other tests.
Conclusions
A new engineered biocatalyst was constructed, with design features specific for ethanol production from dairy waste. To our knowledge, this is the first report showing ethanol production from WP and CWP by engineered E. coli. The strain, selected by a growth/fermentation screening assay, is amenable for further genetic modifications, e.g., gene knockout and heterologous gene expression optimization, to disrupt competing pathways and improve lactose-to- ethanol flux. Our biocatalyst could efficiently ferment two dairy waste streams, derived from an existing valorization chain, without nutritional supplements, providing promising results towards the green, sustainable and economically attractive conversion of such waste into a biofuel.
Methods
Strains and growth media
TOP10 (Invitrogen) E. coli were used for cloning according to manufacturer’s instructions. Bacterial hosts for ethanol production are described in Table 1. All of them were cultured at 37 °C in LB medium (10 g/l NaCl, 10 g/l bactotryptone, 5 g/l yeast extract, and 15 g/l of agar if preparing LB agar plates) following the instructions provided by DSM, ATCC or CGSC. A glycerol stock, routinely stored at -80 °C, was prepared for each strain by mixing 250 μl of sterile 80% glycerol with 750 μl of bacterial culture.
For lactose-supplemented LB, lactose (L2643, Sigma Aldrich) was dissolved in deionized water, filter-sterilized (0.2 μm), and added to autoclaved LB to reach a final concentration of 40 or 80 g/l. When required, 100 mM of phosphate buffer at pH 7.0 was added [46].
WP and CWP were retrieved from the Recetto (Italy) whey processing plant (Negri Alimenti company, Italy). WP comes from a whey protein concentrates extraction process via ultrafiltration. CWP is the result of either a reverse osmosis process on WP, or a ultrafiltration process on whey that has been previously concentrated by reverse osmosis. They were stored in a refrigerator in the plant and transported in non-refrigerated conditions. After delivery, the batches used in this work had an initial lactose concentration and a pH of 42–52 g/l and 6.5–6.7 (WP), and 106–158 g/l and 6.0–6.2 (CWP), respectively. Glucose and galactose, also measured in preliminary tests, typically accounted for only 2% of the total sugars in WP and CWP. Unless differently stated, WP and CWP were stored at -20 °C and filter-sterilized (0.2 μm) before use. When required, 200 mM of piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer at pH 7.0 was added. Antibiotics were always added to maintain plasmids in engineered strains during cloning and fermentation experiments: ampicillin (100 mg/l), kanamycin (50 mg/l) or chloramphenicol (12.5 mg/l).
Cloning
The pdc and adhB gene sequences were designed with the Mr Gene GmbH (Germany) codon-optimization service and obtained via de-novo DNA synthesis. Their sequences were submitted to the MIT Registry of Standard Biological Parts (Registry, http://partsregistry.org) open source archive as BBa_K173016 and BBa_K173017 entries. All the other DNA parts were retrieved from the iGEM 2008 and 2009 DNA Distributions. The pL13 plasmid was assembled via BioBrick Standard Assembly as previously described [42], using the pSB4C5 low-copy number vector backbone. Its construction process and final sequence can be accessed in the BBa_K173022 entry of the Registry. Unless differently stated, competent cells for the candidate strains were prepared as follows: 5 ml of LB were inoculated with glycerol stock of the desired strain and incubated overnight at 37 °C, 220 rpm; the culture was 250-fold diluted in 50 ml of LB in a flask and incubated in the same conditions as above until it reached an OD600 of 0.14 (relative to 200 μl of culture in a 96-well microplate measured via an Infinite F200 reader - Tecan); the culture was chilled in ice and centrifuged (4000 rpm, 4 °C, 15 min); the supernatant was removed and the pellet was resuspended with 30 ml of an MgCl2 (80 mM) + CaCl2 (20 mM) buffer; cells were centrifuged as before and the pellet was resuspended with 2 ml of a CaCl2 (100 mM) + glycerol (1.5%) buffer; cells were transferred into 0.5 ml tubes and stored at -80 °C before use. Bacterial transformation was carried out by heat shock at 42 °C to obtain the recombinant strains.
Growth assays in 96-well microplate
Two to five ml of LB medium were inoculated with the glycerol stocks of the non-engineered and engineered strains. Cultures were incubated overnight at 30 °C, 220 rpm. These cultures were 100-fold diluted in a 96-well microplate in 200 μl of filter-sterilized (0.2 μm) WP, which had been stored at +4 °C for 24 h after its delivery. WP without bacteria was also added to the microplate wells. The microplate was incubated at 30 °C for 21 h in the Infinite F200 (Tecan) reader, with the following kinetic cycle, programmed via the i-control software (Tecan) [52]: linear shaking for 15 s at 3-mm, wait for 5 s, absorbance measurement at 600 nm, repeat every 5 min. At least two independent replicates were analyzed.
Fermentation experiments in 15-ml tubes
Two ml of LB + 40 g/l lactose were inoculated with the glycerol stocks of the strains. Cultures were incubated overnight at 30 °C or 37 °C, 220 rpm. These cultures were centrifuged, their supernatant was removed, the pellet was resuspended in 9 ml of WP + PIPES, and incubated in the same conditions as above for 72 or 168 h. Finally, the cultures were centrifuged, the supernatant was filter-sterilized (0.2 μm) and stored at -20 °C before HPLC analysis. Fermentations using LB + phosphate buffer and 40 g/l or 80 g/l lactose were carried out analogously (at 30 °C, 72 h), with the exceptions that the 2 ml culture was in LB + phosphate buffer + 40 g/l lactose and it was 100-fold diluted in 9 ml of fermentation medium.
Fermentation experiments in pH-controlled bioreactor
Seventy ml of LB + 40 g/l lactose were inoculated with the glycerol stocks of the strains. Cultures were incubated overnight at 30 °C or 37 °C, 220 rpm. These cultures were centrifuged (4000 rpm, 10 min), their supernatant was removed, the pellet was resuspended in 300 ml of fermentation medium, and incubated in the Minifor (Lambda) laboratory-scale bioreactor (0.4-liter vessel) at the same temperature as above, pH 6.6 or 7.0 and agitation set to 4.0 (arbitrary units). The pH was controlled via addition of KOH 3 M, actuated via the PRECIFLOW peristaltic pump (Lambda). Fermentation in 2.4 l culture volume was carried out as above, except that the pre-inoculum was done in 500 ml of LB + 40 g/l lactose, the 7-liter vessel was used, and agitation was set to 6.0 (arbitrary units). Fermentation in LB + 80 g/l lactose was carried out as above, except that the pre-inoculum was done in 3 ml of LB + 40 g/l lactose, and the grown culture was used to inoculate 300 ml of LB + 80 g/l lactose as fermentation medium. Fermentation was carried out in a sterile setup, following the manufacturer recommendations. Gas was allowed to escape from the vessel, to avoid excessive pressure, via a silicon tube connected with a 0.2 μm filter and put into a flask filled with water to limit ethanol loss due to evaporation. The ethanol loss in the described fermentation setup was experimentally demonstrated to be negligible (see Additional file 1: Figure S4).
Quantitative measurements of fermentation performance
Lactose and ethanol concentrations were measured via a Shimadzu 10 AD/vp HPLC system equipped with a Supelco C-610H 30 cm x 7.8 mm column (59320-U, Sigma Aldrich) and a RID 10A detector (Shimadzu). The column was kept at 30 °C. H3PO4 0.1% was used as mobile phase at the flow rate of 0.5 ml/min. The injection of 25 μl was carried out via automatic injector. The pdc and adhB activities were measured via specific enzymatic assays as previously described [53], except that the results were normalized by the activity in the ML308 strain, which was included in each experiment, to decrease the variability of the assay in different reaction mix batches.
Data analysis
In microplate growth assays, for each time point the raw absorbance of WP was subtracted from the absorbance of wells with bacterial cultures to yield the optical density at 600 nm time series of the bacterial cells (OD600, which is proportional to cell density). Doubling time was computed as described by Mandell et al. [54]. One-sided t-tests were performed with Microsoft Excel.
The LabSolutions software (Shimadzu) was used to analyze HPLC data. Lactose and ethanol concentrations were used to compute the fermentation yield as 100*(g/l of ethanol)/(0.54*g/l of lactose), where 0.54 is the theoretical maximum ethanol yield that can be obtained from lactose [46]. Fermentation time (tf) was reported as the time in which residual lactose is less than 1% of the initial lactose. Volumetric productivity was computed as the ethanol concentration at t = tf divided by tf.
Abbreviations
- ATCC:
-
American type culture collection
- CGSC:
-
Coli genetic stock center
- CWP:
-
Concentrated whey permeate
- DSMZ:
-
Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH
- HPLC:
-
High-performance liquid chromatography
- OD600:
-
Optical density at 600 nm
- RBS:
-
Ribosome binding site
- RID:
-
Refraction index detector
- WP:
-
Whey permeate
References
Kosikowski FV. Whey utilization and whey products. J Dairy Sci. 1979;62:1149–60.
Guimaraes PMR, Teixeira JA, Domingues L. Fermentation of lactose to bioethanol by yeasts as part of integrated solutions for the valorisation of cheese whey. Biotechnol Adv. 2010;28:375–84.
Siso MIG. The biotechnological utilization of cheese whey: a review. Bioresour Technol. 1996;57(1):1–11.
Prazeres AR, Carvalho F, Rivas J. Cheese whey management: A review. J Environ Manag. 2012;110:48–68.
Sansonetti S, Curcio S, Calabro V, Iorio G. Bioethanol production by fermentation of ricotta cheese whey as an effective alternative non-vegetable source. Biomass Bioenergy. 2009;33(12):1687–92.
De Wit JN. Lecturer’s Handbook on Whey and Whey Products. 1st ed. Brussels: European Whey Products Association; 2001.
Ling KC. Whey to ethanol: a biofuel role for dairy cooperatives? Washington DC: USDA Rural Development; 2008.
Jarboe LR, Zhang X, Wang X, Moore JC, Shanmugam KT, Ingram LO. Metabolic engineering for production of biorenewable fuels and chemicals: Contributions of synthetic biology. J Biomed Biotechnol. 2010;2010:761042.
Tomaszewska M, Bialonczyk L. Ethanol production from whey in a bioreactor coupled with direct contact membrane distillation. Catal Today. 2016;268:156–63.
De Glutz FN. Fuel bioethanol production from whey permeate. PhD Thesis, Ecole Polytechnique Federale de Lausanne, 2009.
Gancedo JM. Yeast carbon catabolite repression. Microbiol Mol Biol Rev. 1998;62(2):334–61.
Zimmermann FK, Scheel I. Mutants of Saccharomyces cerevisiae resistant to carbon catabolite repression. Mol Gen Genet. 1977;154(1):75–82.
Silveira WB, Passos F, Mantovani HC, Passos FML. Ethanol production from cheese whey permeate by Kluyveromyces marxianus UFV-3: a flux analysis of oxido-reductive metabolism as a function of lactose concentration and oxygen levels. Enzyme Microbiol Technol. 2005;36:930–6.
Vincenzi A, Maciel MJ, Burlani EL, Oliveira EC, Volpato G, Lehn DN, de Souza CFV. Ethanol bio-production from ricotta cheese whey by several strains of the yeast Kluyveromyces. Am J Food Technol. 2014;9(6):281–91.
Dien BS, Cotta MA, Jeffries TW. Bacteria engineered for fuel ethanol production: current status. Appl Microbiol Biotechnol. 2003;63(3):258–66.
Ingram LO, Conway T, Clark DP, Sewell GW, Preston JF. Genetic engineering of ethanol production in Escherichia coli. Appl Environ Microbiol. 1987;53(10):2420–5.
Ingram LO, Conway T. Expression of Different Levels of Ethanologenic enzymes from Zymomonas mobilis in recombinant strains of Escherichia coli. Appl Environ Microbiol. 1988;54(2):397–404.
Alterthum F, Ingram LO. Efficient ethanol production from glucose, lactose, and xylose by recombinant Escherichia coli. Appl Environ Microbiol. 1989;55(8):1943–8.
Ohta K, Beall DS, Mejia JP, Shanmugam KT, Ingram LO. Genetic improvement of Escherichia coli for ethanol production: chromosomal integration of Zymomonas mobilis genes encoding Pyruvate Decarboxylase and Alcohol Dehydrogenase II. Appl Environ Microbiol. 1991;57(4):893–900.
Lawford HG, Rousseau JD. Loss of ethanologenicity in Escherichia coli B recombinants pLOI297 and KO11 during growth in the absence of antibiotics. Biotechnol Lett. 1995;17(7):751–6.
Asghari A, Bothast RJ, Doran JB, Ingram LO. Ethanol production from hemicellulose hydrolysates of agricultural residues using genetically engineered Escherichia coli strain KO11. J Ind Microbiol. 1996;16:42–7.
Moniruzzaman M, Dien BS, Ferrer B, Hespell RB, Dale BE, Ingram LO, Bothast RJ. Ethanol production from AFEX pretreated corn fiber by recombinant bacteria. Biotechnol Lett. 1996;18:985–90.
Martinez A, York SW, Yomano LP, Pineda VL, Davis FC, Shelton JC, Ingram LO. Biosynthetic burden and plasmid burden limit expression of chromosomally integrated heterologous genes (pdc, adhB) in Escherichia coli. Biotechnol Prog. 1999;15(5):891–7.
Orencio-Trejo M, Flores N, Escalante A, Hernandez-Chavez G, Bolivar F, Gosset G, Martinez A. Metabolic regulation analysis of an ethanologenic Escherichia coli strain based on RT-PCR and enzymatic activities. Biotechnol Biofuels. 2008;1(1):8.
Woodruff LB, May BL, Warner JR, Gill RT. Towards a metabolic engineering strain “commons”: an Escherichia coli platform strain for ethanol production. Biotechnol Bioeng. 2013;110(5):1520–6.
Turner PC, Yomano LP, Jarboe LR, York SW, Baggett CL, Moritz BE, Zentz EB, Shanmugam KT, Ingram LO. Optical mapping and sequencing of the Escherichia coli KO11 genome reveal extensive chromosomal rearrangements, and multiple tandem copies of the Zymomonas mobilis pdc and adhB genes. J Ind Microbiol Biotechnol. 2012;39(4):629–39.
Zhou B, Martin GJ, Pamment NB. A novel assay for rapid in vivo determination of phenotypic stability of recombinant ethanol-producing microorganisms. Bioresour Technol. 2009;100(11):2823–7.
Dumsday GJ, Zhou B, Yaqin W, Stanley GA, Pamment NB. Comparative stability of ethanol production by Escherichia coli KO11 in batch and chemostat culture. J Ind Microbiol Biotechnol. 1999;23:701–8.
Underwood SA, Buszko ML, Shanmugam KT, Ingram LO. Flux through citrate synthase limits the growth of ethanologenic Escherichia coli KO11 during xylose fermentation. Appl Environ Microbiol. 2002;68(3):1071–81.
Yomano LP, York SW, Zhou S, Shanmugam KT, Ingram LO. Re-engineering Escherichia coli for ethanol production. Biotechnol Lett. 2008;30:2097–103.
Wargacki AJ, Leonard E, Win MN, Regitsky DD, Santos CN, Kim PB, Cooper SR, Raisner RM, Herman A, Sivitz AB, et al. An engineered microbial platform for direct biofuel production from brown macroalgae. Science. 2012;335(6066):308–13.
Guimaraes WV, Dudey GL, Ingram LO. Fermentation of sweet whey by ethanologenic Escherichia coli. Biotechnol Bioeng. 1992;40:41–5.
Leite AR, Guimaraes WV, de Araujo EF, Silva DO. Fermentation of sweet whey by recombinant Escherichia coli KO11. Braz J Microbiol. 2000;31:212–5.
Fernandez-Sandoval MT, Huerta-Beristain G, Trujillo-Martinez B, Bustos P, Gonzalez V, Bolivar F, Gosset G, Martinez A. Laboratory metabolic evolution improves acetate tolerance and growth on acetate of ethanologenic Escherichia coli under non-aerated conditions in glucose-mineral medium. Appl Microbiol Biotechnol. 2012;96(5):1291–300.
Trinh CT, Unrean P, Srienc F. Minimal Escherichia coli cell for the most efficient production of ethanol from hexoses and pentoses. Appl Environ Microbiol. 2008;74(12):3634–43.
Hespell RB, Wyckoff H, Dien BS, Bothast RJ. Stabilization of pet operon plasmids and ethanol production in Escherichia coli strains lacking lactate dehydrogenase and pyruvate formate-lyase activities. Appl Environ Microbiol. 1996;62:4594–7.
Dien BS, Nichols NN, O’Bryan PJ, Bothast RJ. Development of new ethanologenic Escherichia coli strains for fermentation of lignocellulosic biomass. Appl Biochem Biotechnol. 2000;84–86:181–96.
Kim Y, Ingram LO, Shanmugam KT. Construction of an Escherichia coli K-12 mutant for homoethanologenic fermentation of glucose or xylose without foreign genes. Appl Environ Microbiol. 2007;73(6):1766–71.
Zhou S, Iverson AG, Grayburn WS. Engineering a native homoethanol pathway in Escherichia coli B for ethanol production. Biotechnol Lett. 2008;30:335–42.
Politi N, Pasotti L, Zucca S, Casanova M, Micoli G, Cusella De Angelis MG, Magni P. Half-life measurements of chemical inducers for recombinant gene expression. J Biol Eng. 2014;8:5.
Weiss R. Cellular computation and communications using engineered genetic regulatory networks. Ph.D. thesis, Massachusetts Institute of Technology. 2001.
Shetty R, Endy D, Knight T. Engineering BioBrick vectors from BioBrick parts. J Biol Eng. 2008;2:5.
Zucca S, Pasotti L, Politi N, Cusella De Angelis MG, Magni P. A standard vector for the chromosomal integration and characterization of BioBrickTM parts in Escherichia coli. J Biol Eng. 2013;7:12.
Pasotti L, Zucca S. Advances and computational tools towards predictable design in biological engineering. Comput Math Methods Med. 2014;2014:369681. doi:10.1155/2014/369681.
Tian T, Salis HM. A predictive biophysical model of translational coupling to coordinate and control protein expression in bacterial operons. Nucleic Acids Res. 2015;43(14):7137–51.
Beall DS, Ohta K, Ingram LO. Parametric studies of ethanol production from xylose and other sugars by recombinant Escherichia coli. Biotechnol Bioeng. 1991;38(3):296–303.
Massaiu I, Pasotti L, Casanova M, Politi N, Zucca S, Cusella De Angelis MG, Magni P. Quantification of the gene silencing performances of rationally designed synthetic small RNAs. Syst Synth Biol. 2015;9(3):107–23.
Kelly JR. Tools and reference standards supporting the engineering and evolution of synthetic biological systems. Ph.D. thesis, Massachusetts Institute of Technology. 2008.
Casanova M, Pasotti L, Zucca S, Politi N, Massaiu I, Calvio C, Cusella De Angelis MG, Magni P. A BioBrickTM-compatible vector for allelic replacement using the xylE gene as selection marker. Biological Procedures Online. 2016;18:6.
Archer CT, Kim JF, Jeong H, Park JH, Vickers CE, Lee SY, Nielsen LK. The genome sequence of E. coli W (ATCC 9637): comparative genome analysis and an improved genome-scale reconstruction of E. coli. BMC Genomics. 2011;12:9.
Chung CT, Niemela SL, Miller RH. One-step preparation of competent Escherichia coli : transformation and storage of bacterial cells in the same solution. PNAS. 1989;86:2172–5.
Zucca S, Pasotti L, Politi N, Casanova M, Mazzini G, Cusella De Angelis MG, Magni P. Multi-faceted characterization of a novel LuxR-repressible promoter library for Escherichia coli. PLoS One. 2015;10(5):e0126264.
Pasotti L, Zucca S, Casanova M, Politi N, Massaiu I, Mazzini G, Micoli G, Calvio C, Cusella De Angelis MG, Magni P. Methods for genetic optimization of biocatalysts for biofuel production from dairy waste through synthetic biology. Conf Proc IEEE Eng Med Biol Soc. 2015;2015:953–6. doi:10.1109/EMBC.2015.7318521.
Mandell DJ, Lajoie MJ, Mee MT, Takeuchi R, et al. Biocontainment of genetically modified organisms by synthetic protein design. Nature. 2015;518:55–60.
Acknowledgements
The authors want to thank Giacomo Zambianchi, Nicolo Politi, Ilaria Massaiu and Massimo Bellato for their helpful discussions and support in protocol development and fermentation experiments. The authors also want to thank, for their preliminary work, Giulio Chiesa, Elisa Del Fabbro, Rita De Molfetta, Letizia Diamante, Manuel Lupotto, Matteo Meroso, Susanna Schiavi, Andrea Turcato, Valerio Vitelli and Daniela Galli as members of the UNIPV-Pavia iGEM 2009 Team, and Edoardo Mazzocchi and Chiara Pugno as undergraduate students. Finally, the authors want to thank Tommaso Mazzocchi (Polo Tecnologico di Pavia) and Sergio Mantegazza (Negri Alimenti) for their help in dairy waste retrieval. This work was supported by Fondazione Cariplo through the grant 2015-0397 “Conversion of industrial bio-waste into biofuels and bioproducts through synthetic biology”.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request (microplate reader data: Microsoft Excel spreadsheets for growth and enzyme assays; HPLC data: sugar and fermentation product quantifications).
Authors’ contributions
LP, SZ and PM conceived the study, designed the experiments and analyzed the data. LP and SZ constructed the plasmids. LP, SZ and MC carried out growth and fermentation experiments. MC carried out enzymatic assays. SZ, MC and GM carried out measurements via HPLC. LP drafted the manuscript and PM and MGCDA finalized it. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Figure S1.
Fermentation of LB supplemented with 80 g/l of lactose in a pH-controlled bioreactor for W-pL13. Figure S2. Fermentation of WP without filter-sterilization in a pH-controlled bioreactor for W-pL13 in a 2.4-liter culture. Figure S3. Fermentation of CWP in a pH-controlled bioreactor for W-pLOI297. Figure S4. Evaluation of ethanol evaporation in a bioreactor experiment. (PDF 352 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pasotti, L., Zucca, S., Casanova, M. et al. Fermentation of lactose to ethanol in cheese whey permeate and concentrated permeate by engineered Escherichia coli . BMC Biotechnol 17, 48 (2017). https://doi.org/10.1186/s12896-017-0369-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12896-017-0369-y