
Abstract
Background
A possible role for prostate cancer in Lynch syndrome has been debated based on observations of mismatch-repair defective tumors and reports of an increased risk of prostate cancer in mutation carriers. Potential inclusion of prostate cancer in the Lynch syndrome tumor spectrum is relevant for family classification, risk estimates and surveillance recommendations in mutation carriers.
Methods
We used the population-based Danish HNPCC-register to identify all prostate cancers that developed in mutation carriers and in their first-degree relatives from 288 Lynch syndrome families. The tumors were evaluated for clinicopathologic features and mismatch-repair status, and the cumulative risk of prostate cancer was determined.
Results
In total, 28 prostate cancers developed in 16 mutation carriers and in 12 first-degree relatives at a median age of 63 years. The majority of the tumors were high-grade tumors with Gleason scores 8–10. Prostate cancer was associated with mutations in MSH2, MLH1 and MSH6 with loss of the respective mismatch repair protein in 69 % of the tumors, though a MSI-high phenotype was restricted to 13 % of the tumors. The cumulative risk of prostate cancer at age 70 was 3.7 % (95 % CI: 2.3–4.9).
Conclusion
We provide evidence to link prostate cancer to Lynch syndrome through demonstration of MMR defective tumors and an increased risk of the disease, which suggests that prostate cancer should be considered in the diagnostic work-up of Lynch syndrome.
Similar content being viewed by others


Background
Lynch syndrome is a multi-tumor syndrome with the highest risks for colorectal cancer and endometrial cancer though a number of other tumor types, e.g. cancer of the urinary tract, the small bowel and the ventricle, ovarian cancer, brain tumors and skin tumors develop at increased incidence [1, 2]. Other tumor types assumed to represent sporadic tumors in families with hereditary cancer, e.g. breast cancer, pancreatic cancer and sarcoma may indeed develop as part of the syndrome. This is suggested based on identification of mismatch repair (MMR) defective tumors of these subtypes and demonstration of an increased risk of these tumor types in mutation carriers [3–10].
Prostate cancer is the most common tumor type in men in the Western world with an estimated lifetime risk of 18 % and a median age at diagnosis of 67 years [1]. Worldwide, prostate cancer is the sixth common tumor with more than 250,000 deaths annually [11]. In Denmark, prostate cancer constitutes 23 % of all male cancers with an estimated risk of 10 % for disease development before age 75 [12]. The role of prostate cancer in Lynch syndrome is unresolved though molecular investigations and epidemiologic studies have suggested a potential link to the syndrome [1, 8, 13]. The MMR system has been suggested to influence prostate carcinogenesis e.g. through an increased risk of prostate cancer linked to single nucleotide polymorphisms in MLH1 and MSH3, and a role for complex structural rearrangements in MSH2 and MSH6 as a mechanism underlying the hypermutation in aggressive prostate cancer [14–20]. We assessed the role of prostate cancer in the Danish Lynch syndrome cohort with characterization of MMR status and risk estimates.
Methods
Patients and tumor samples
The Danish Hereditary Non-Polyposis Colorectal Cancer (HNPCC) Register is a national Danish register containing all families identified with proven or suspected hereditary cancer. Through research collaborations, data from the register is freely available. We obtained data on all adenocarcinomas of the prostate that had developed in carriers of a disease-predisposing MMR gene mutation in MLH1, MSH2, MSH6 or PMS2 and in their first-degree relatives. Clinical data were obtained from pathology reports and clinical files. All patients provided an informed consent for inclusion into the Danish HNPCC register during genetic counseling sessions. Ethical approval for the study was granted from the Ethical Committee at The Capital Region of Copenhagen, Denmark (H-D-2007–0032). All tumor specimens available were collected for analysis of MMR status. The tumors were pathologically reviewed regarding their Gleason scores and the presence of tumor-infiltrating lymphocytes (TIL) (cut-off ≥4 per high-power field) [8, 21] by a pathologist (PJ), who was blinded to MMR status.
Immunohistochemistry and analysis of microsatellite instability
All tumors were immunohistochemically stained for the MMR proteins MLH1, PMS2, MSH2 and MSH6. Briefly, 4-μm sections were placed on SuperFrost® Plus microscope slides. Antigen retrieval was performed in a pressure boiler in Target Retrieval Solution, pH 9 (Dako, Glostrup, Denmark) and stained in an automated immunostainer (Autostainer Plus, Dako, Glostrup, Denmark) using Dako EnVision™FLEX+ Detection System, Peroxidase/DAB, Rabbit/Mouse (Dako, Glostrup, Denmark), according to the manufacturers' instructions. The antibodies used were MLH1, clone ES05 (Dako, Glostrup, Denmark, dilution 1:100), PMS2, clone A16-4 (BD Pharmingen, San Diego, CA, dilution 1:300), MSH2, clone FE11 (Calbiochem, Merck KgaA, Darmstadt, Germany, dilution 1:100), and MSH6, clone EPR3945 (Epitomics, Burlingame, dilution 1:100). Tumor MMR protein expression was assessed as retained (normal), lost, or reduced (i.e. tumor cell staining intensity was reduced compared with that of the normal internal control).
For analysis of microsatellite instability (MSI), non-necrotic tumor areas were macro-dissected from the paraffin-embedded tumor blocks. DNA extraction was performed from three 5-mm sections using the Qiagen FFPE Kit (Qiagen Valencia, CA) according to the manufacturer’s instructions. DNA concentration was determined using a Qubit Fluorometric Quantitation (Invitrogen) and the products run on a 3130XL Genetic Analyzer (Applied Biosystems, Foster City, CA). The analysis was performed using the MSI Analysis System, Version 1.2 (Promega, Madison, WI) and included the 5 mononucleotide markers BAT-25 BAT-26, NR-21, NR-24, and MONO-27 (Promega, MSI Analysis System, Version 1.2, Madison, WI). The results were evaluated using GeneMapper Software Version 4.0 (Applied Biosystems, Foster City, CA) and defined as MSI high when ≥2 markers were unstable, MSI low when one marker was unstable and MSS when none of the markers were unstable.
Statistical analysis
Genotypic and phenotypic data from all mutation carriers and their first-degree relatives were transferred into R i386 3.1.0 (R: A Language and Environment for Statistical Computing, 2011, R Foundation for Statistical Computing, Vienna, Austria). EPCAM mutations (identified in one family) were pooled together with MSH2 mutations, while 7 PMS2 mutation families (none of which contained any prostate cancers) were excluded from the analyses. Mutation carriers were weighted by 1 and first-degree relatives by 0.5 motivated by a 50 % risk of carrying the inherited mutation. The event times used were age at diagnosis, age at death or current age (censored at May 14, 2014). Cumulative incidences were calculated with death as a competing risk (cmprsk: Subdistribution Analysis of Competing Risks, 2011, Bob Gray, R package version 2.2-2). Confidence intervals were calculated at age 70 using a non-parametric bootstrap. Permutation tests with 10,000 replicates were used to calculate p-values with significance set at p < 0.05.
Results
In total, 288 Lynch syndrome families with disease-predisposing germline mutations in MLH1, MSH2, MSH6 or PMS2 were identified in the Danish HNPCC register. In this cohort of 1609 males (677 mutation carriers and 932 first-degree relatives), prostate cancer developed in 16 mutation carriers and in 12 first-degree relatives. The median age at diagnosis of prostate cancer was 61 (range 52–78) years for the mutation carriers and 63 (range 53–81) years for the first-degree relatives. All tumors were adenocarcinomas with Gleason scores between 6 and 10. The tumors were linked to disease-predisposing mutations in MSH2 (n = 14), MLH1 (n = 8) and MSH6 (n = 6) (Table 1). Among the 28 men diagnosed with prostate cancer, 16 had a previous cancer diagnosis, which included colon cancer in 15 cases. Four prostate cancers had developed among the 593 male non-mutation carriers (0.67 %) compared to 2.22 % of the mutation carriers and 1.39 % of the first-degree relatives.
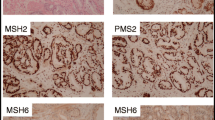
Tumor tissue could be retrieved for MMR analysis from 16 tumors (derived from 10 mutation carriers and 6 first-degree relatives), with loss of expression for the respective MMR proteins in 11/16 tumors (including 7/10 tumors from mutation carriers (Table 1). Notably, MMR protein loss was detected in all MSH2 and MSH6 associated tumors prostate cancers. MSI analysis applied standard diagnostic markers and revealed a MSI-high phenotype in 2 tumors, a MSI-low phenotype in 6 tumors and a MSS phenotype in 8 tumors (Table 1, Fig. 1). Notably, all MLH1-associated tumors has a microsatellite stable phenotype. Pathologic review revealed TIL in 12/16 tumors, including all MMR defective tumors. Gleason scores tended to be high in MMR defective prostate cancer with Gleason scores of 8–10 in 7/11 MMR defective prostate cancers (Table 1).

a A prostate cancer from an individual with a MSH2 mutation showing normal expression for MLH1 and PMS2 (A and B) and loss of expression for MSH2 and MSH6 (C and D); b microsatellite instability for the markers BAT-26, NR-21, BAT-25, NR-24 and MONO-27 in the same prostate cancer
Risk analysis could be performed based on 1488/1609 males from whom complete data were available. The cumulative risk for prostate cancers at age 70 was 3.7 % (95 % CI: 2.32–4.92) in mutation carriers and first-degree relatives compared to 593 for non-mutation carriers in these families (Fig. 2a). No significant differences could be demonstrated in relation to disease-predisposing gene; MLH1 4.4 % (95 % CI: 1.44; 7.04), MSH2 3.9 % (95 % CI: 1.96–5.70) and MSH6 2.5 % (95 % CI: 0.56–4.12) (Fig. 2b).

Non-parametric risk estimates showing a age-specific cumulative risks for prostate cancer; b mortality rates in MLH1, MSH2 and MSH6 families
Discussion
In the Danish Lynch syndrome cohort, 28 prostate cancers were identified. These tumors were diagnosed at a median age of 63 years, which is in line with reports of prostate cancers diagnosed at median 59–65 years in Lynch syndrome [9, 22–26]. MMR protein loss in line with the underlying MMR gene mutation was identified in 11/16 prostate cancers, including all MSH2 and MSH6 mutant tumors and supports observations of a high degree, 69–100 %, of MMR deficiency in prostate cancers in Lynch syndrome [8, 24, 27]. MSH2 mutations were found in 14/28 prostate cancers and this MMR gene has been linked to an expanded spectrum of extracolonic tumors with an increased risk for e.g. urothelial cancer, brain tumors and skin tumors [6, 8–10, 22–24, 28, 29]. A MSI-high phenotypes was identified only in 2 prostate cancers using a standard MSI markers panel with an additional number of tumors showing a MSI-low phenotype. This observation supports an earlier report of MSI defects in 4–10 % of Lynch syndrome associated prostate cancers [30]. MSI phenotypes were predominantly observed in MSH2 and MSH6 associated tumors and could potentially reflect alternate mechanisms by which MSI is acquired in prostate cancer or an association with tumor differentiation [19] [31]. These observations support a role for MMR defects in prostate carcinogenesis and link germline MMR defects to the development of prostate cancer.
MMR-defective tumors show histopathologic characteristics that include poor differentiation and lymphocytic reactions with an increased number of TIL [8]. Blinded analysis of TIL showed a striking correlation with MMR defects with TIL in all MMR defective prostate cancer and in only 1/5 MSS and MMR proficient prostate cancers. TIL has been suggested to represent an adverse prognostic factor in prostate cancer [32–34]. Of the 11 MMR defective prostate cancers in our study, 7 had a Gleason score of ≥8 suggesting aggressive tumors. Hereditary prostate cancers, also associated with BRCA2 mutations, have been suggested to have an accelerated tumor development an aggressive phenotype. Knowledge about prostate cancer in Lynch syndrome is scarce, but early age at onset, frequent TIL and an aggressive phenotype warrants further investigation related to a possible role for surveillance and potential therapeutic implications from e.g. immunotherapy with PD-1 inhibitors [35].
The cumulative risk of prostate cancer at age 70 was 3.7 % in mutation carriers. No significant differences were discerned in relation to disease-predisposing gene, but this analysis is based on very limited numbers. Under the assumption that MMR-defective prostate cancer signifies Lynch syndrome, mutation carriers can be estimated to be at a 2- to 3-fold increased risk of prostate cancer compared to the general population [1, 13]. Growing data suggest that MMR defects in prostate cancer may signify chromoplexy, whereby a single hit infers genetic complexity of relevance for prostate cancer initiation, progression and therapeutics [18, 19].
Conclusions
The Danish Lynch syndrome cohort contains 28 prostate cancers that developed at a median age of 63 years, showed high Gleason scores and frequent TILs. The tumors were predominantly linked to MSH2 mutations. Frequent MMR defects consistent with the underlying germline defects suggest that prostate cancer is included in Lynch syndrome tumor spectrum and should be considered during genetic counseling.
Availability of data and materials
All available data from the cases included are summarized in Table 1. Additional data on these individuals and their families (e.g. detailed mutation data and family data) are freely available from the Danish HNPCC-register though contact with the principal investigator, Mef.Nilbert@regionh.dk.
Abbreviations
- CI:
-
confidence interval
- HNPCC:
-
hereditary nonpolyposis colorectal cancer
- MMR:
-
mismatch repair
- MSI:
-
microsatellite instability
- MSS:
-
microsatellite stable
- TIL:
-
tumor-infiltrating lymphocytes.
References
Raymond VM, Mukherjee B, Wang F, Huang SC, Stoffel EM, Kastrinos F, Syngal S, Cooney KA, Gruber SB. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013;31(14):1713–8.
Win AK, Lindor NM, Winship I, Tucker KM, Buchanan DD, Young JP, Rosty C, Leggett B, Giles GG, Goldblatt J, et al. Risks of colorectal and other cancers after endometrial cancer for women with Lynch syndrome. J Natl Cancer Inst. 2013;105(4):274–9.
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–30.
Nilbert M, Therkildsen C, Nissen A, Akerman M, Bernstein I. Sarcomas associated with hereditary nonpolyposis colorectal cancer: broad anatomical and morphological spectrum. Fam Cancer. 2009;8(3):209–13.
Walsh MD, Buchanan DD, Cummings MC, Pearson SA, Arnold ST, Clendenning M, Walters R, McKeone DM, Spurdle AB, Hopper JL, et al. Lynch syndrome-associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin Cancer Res. 2010;16(7):2214–24.
van der Post RS, Kiemeney LA, Ligtenberg MJ, Witjes JA, Hulsbergen-van de Kaa CA, Bodmer D, Schaap L, Kets CM, van Krieken JH, Hoogerbrugge N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. 2010;47(7):464–70.
da Silva FC, de Oliveira LP, Santos EM, Nakagawa WT, Aguiar Junior S, Valentin MD, Rossi BM, de Oliveira Ferreira F. Frequency of extracolonic tumors in Brazilian families with Lynch syndrome: analysis of a hereditary colorectal cancer institutional registry. Fam Cancer. 2010;9(4):563–70.
Rosty C, Walsh MD, Lindor NM, Thibodeau SN, Mundt E, Gallinger S, Aronson M, Pollett A, Baron JA, Pearson S, et al. High prevalence of mismatch repair deficiency in prostate cancers diagnosed in mismatch repair gene mutation carriers from the colon cancer family registry. Fam Cancer. 2014;13(4):573–82.
Haraldsdottir S, Hampel H, Wei L, Wu C, Frankel W, Bekaii-Saab T, de la Chapelle A, Goldberg RM. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16(7):553–7.
Joost P, Therkildsen C, Dominguez-Valentin M, Jonsson M, Nilbert M. Urinary Tract Cancer in Lynch Syndrome; Increased Risk in Carriers of MSH2 Mutations. Urology. 2015.
Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127(12):2893–917.
Engholm G, Ferlay J, Christensen N, Bray F, Gjerstorff ML, Klint A, Kotlum JE, Olafsdottir E, Pukkala E, Storm HH. NORDCAN--a Nordic tool for cancer information, planning, quality control and research. Acta Oncol. 2010;49(5):725–36. Available from http://www.ancr.nu, accessed on 02/12/2015.
Ryan S, Jenkins MA, Win AK. Risk of prostate cancer in Lynch syndrome: a systematic review and meta-analysis. Cancer Epidemiol Biomarkers Prev. 2014;23(3):437–49.
Langeberg WJ, Kwon EM, Koopmeiners JS, Ostrander EA, Stanford JL. Population-based study of the association of variants in mismatch repair genes with prostate cancer risk and outcomes. Cancer Epidemiol Biomarkers Prev. 2010;19(1):258–64.
Tanaka Y, Zaman MS, Majid S, Liu J, Kawakami K, Shiina H, Tokizane T, Dahiya AV, Sen S, Nakajima K. Polymorphisms of MLH1 in benign prostatic hyperplasia and sporadic prostate cancer. Biochem Biophys Res Commun. 2009;383(4):440–4.
Hirata H, Hinoda Y, Kawamoto K, Kikuno N, Suehiro Y, Okayama N, Tanaka Y, Dahiya R. Mismatch repair gene MSH3 polymorphism is associated with the risk of sporadic prostate cancer. J Urol. 2008;179(5):2020–4.
Jafary F, Salehi M, Sedghi M, Nouri N, Jafary F, Sadeghi F, Motamedi S, Talebi M. Association between mismatch repair gene MSH3 codons 1036 and 222 polymorphisms and sporadic prostate cancer in the Iranian population. Asian Pac J Cancer Prev. 2012;13(12):6055–7.
Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013;153(3):666–77.
Pritchard CC, Morrissey C, Kumar A, Zhang X, Smith C, Coleman I, Salipante SJ, Milbank J, Yu M, Grady WM, et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun. 2014;5:4988.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18(1):11–22.
Young J, Simms LA, Biden KG, Wynter C, Whitehall V, Karamatic R, George J, Goldblatt J, Walpole I, Robin SA, et al. Features of colorectal cancers with high-level microsatellite instability occurring in familial and sporadic settings: parallel pathways of tumorigenesis. Am J Pathol. 2001;159(6):2107–16.
Win AK, Lindor NM, Young JP, Macrae FA, Young GP, Williamson E, Parry S, Goldblatt J, Lipton L, Winship I, et al. Risks of primary extracolonic cancers following colorectal cancer in lynch syndrome. J Natl Cancer Inst. 2012;104(18):1363–72.
Goecke T, Schulmann K, Engel C, Holinski-Feder E, Pagenstecher C, Schackert HK, Kloor M, Kunstmann E, Vogelsang H, Keller G, et al. Genotype-phenotype comparison of German MLH1 and MSH2 mutation carriers clinically affected with Lynch syndrome: a report by the German HNPCC Consortium. J Clin Oncol. 2006;24(26):4285–92.
Grindedal EM, Moller P, Eeles R, Stormorken AT, Bowitz-Lothe IM, Landro SM, Clark N, Kvale R, Shanley S, Maehle L. Germ-line mutations in mismatch repair genes associated with prostate cancer. Cancer Epidemiol Biomarkers Prev. 2009;18(9):2460–7.
Pande D, Negi R, Karki K, Dwivedi US, Khanna RS, Khanna HD. Simultaneous progression of oxidative stress, angiogenesis, and cell proliferation in prostate carcinoma. Urol Oncol. 2013;31(8):1561–6.
Engel C, Loeffler M, Steinke V, Rahner N, Holinski-Feder E, Dietmaier W, Schackert HK, Goergens H, von Knebel Doeberitz M, Goecke TO, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol. 2012;30(35):4409–15.
Bauer CM, Ray AM, Halstead-Nussloch BA, Dekker RG, Raymond VM, Gruber SB, Cooney KA. Hereditary prostate cancer as a feature of Lynch syndrome. Fam Cancer. 2011;10(1):37–42.
Vasen HF, Stormorken A, Menko FH, Nagengast FM, Kleibeuker JH, Griffioen G, Taal BG, Moller P, Wijnen JT. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol. 2001;19(20):4074–80.
Kastrinos F, Stoffel EM, Balmana J, Steyerberg EW, Mercado R, Syngal S. Phenotype comparison of MLH1 and MSH2 mutation carriers in a cohort of 1,914 individuals undergoing clinical genetic testing in the United States. Cancer Epidemiol Biomarkers Prev. 2008;17(8):2044–51.
Ahman AK, Jonsson BA, Damber JE, Bergh A, Gronberg H. Low frequency of microsatellite instability in hereditary prostate cancer. BJU Int. 2001;87(4):334–8.
Chen Y, Wang J, Fraig MM, Metcalf J, Turner WR, Bissada NK, Watson DK, Schweinfest CW. Defects of DNA mismatch repair in human prostate cancer. Cancer Res. 2001;61(10):4112–21.
Ness N, Andersen S, Valkov A, Nordby Y, Donnem T, Al-Saad S, Busund LT, Bremnes RM, Richardsen E. Infiltration of CD8+ lymphocytes is an independent prognostic factor of biochemical failure-free survival in prostate cancer. Prostate. 2014;74(14):1452–61.
Flammiger A, Bayer F, Cirugeda-Kuhnert A, Huland H, Tennstedt P, Simon R, Minner S, Bokemeyer C, Sauter G, Schlomm T, et al. Intratumoral T but not B lymphocytes are related to clinical outcome in prostate cancer. APMIS. 2012;120(11):901–8.
Karja V, Aaltomaa S, Lipponen P, Isotalo T, Talja M, Mokka R. Tumour-infiltrating lymphocytes: A prognostic factor of PSA-free survival in patients with local prostate carcinoma treated by radical prostatectomy. Anticancer Res. 2005;25(6C):4435–8.
Castro E, Goh C, Olmos D, Saunders E, Leongamornlert D, Tymrakiewicz M, Mahmud N, Dadaev T, Govindasami K, Guy M, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31(14):1748–57.
Acknowledgments
Financial support was granted from the Swedish Cancer Society, the Danish Cancer Society, the Nilsson Cancer Fund and the Kamprad Cancer Fund and the ALF Funds at the Lund University Medical Faculty.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have read and approved of the final version of the manuscript. MDV: Protocol/project development, experimental analysis, data collection and analysis, manuscript writing/editing. PJ: Protocol/project development, data analysis, manuscript writing/editing. CT: Data collection or management, data analysis, manuscript writing/editing. MJ: Experimental support, manuscript writing/editing. ER: Experimental support, manuscript writing/editing. MN: Protocol/project development, data management, manuscript editing.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dominguez-Valentin, M., Joost, P., Therkildsen, C. et al. Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol 16, 15 (2016). https://doi.org/10.1186/s12894-016-0130-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12894-016-0130-1