
Abstract
Background
Neurofibromatosis comprises neurofibromatosis type 1 (NF1) and type 2 (NF2). Major tumor type of NF1 are neurofibroma recognized as benign peripheral nerve tumor, malignant peripheral nerve sheath tumor (MPNST), and glioma.
Case presentation
We report a woman with a special condition, whose tumors in body surfaces were benign neurofibroma and tumors in posterior mediastinum are MPNST. The chest-enhanced CT suggested a round soft tissue density in posteriormediastium. The diagnosis was established by pathology and immunohistochemistry. A single-stage thoracoscopic mediastinal mass resection was performed. The whole operation went smoothly and the CT scan of lungs did not show relapse of tumor three months later.
Conclusions
The appearance of neurofibroma should draw particular attention to the possibility of developing MPNST. More careful imaging examinations should be carried out, and pathological examination could diagnose it.
Similar content being viewed by others

Background
Neurofibromatosis is a autosomal dominant disease that mainly implicates nervous system and causes tumor with distinct predisposition [1]. According to the classification of the National Institute of Health (NIH) in 1988, neurofibromatosis comprises neurofibromatosis type 1 (NF1) and type 2 (NF2). Major tumor type of NF1 are neurofibroma recognized as benign peripheral nerve tumor, malignant peripheral nerve sheath tumor (MPNST), and glioma [1,2,3,4]. NF2 includes bilateral vestibular nerve schwannomas as the most common tumor type, meningiomas, and ependymomas [1, 5].
From clinical experience, neurocutaneous tumors rarely associated with esophageal neurogenic tumors and related symptoms. The patient reported in this case is a 50-year-old woman with a special condition, whose tumors in body surfaces were benign neurofibroma (Fig. 1) and tumors in posterior mediastinum are MPNST. Despite neurofibroma and nerve sheath tumor are both Schwann cell-derived tumors, their tumor characteristics and clinical manifestations are significantly different.

Photograph of neurofibroma in body surface
Case presentation
This study was conducted under approval of the Ethics Committee of First Hospital of Jilin University, and the protocol was accorded with its standards.
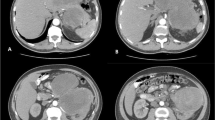
This patient was a 50-year-old woman admitted to our department due to a posterior mediastinal mass detected by computed tomography (CT) scan 7 days ago. The symptom of oppression in chest did not improve after anti-inflammatory treatment. At admission, her blood pressure, pulse rate, respiratory rate and body temperature were all in the normal range. She had no cough and expectoration, no fever, no significant chest and back pain, and no muscle weakness. She had no hypertension, heart disease, diabetes, any infectious disease or drug allergy history. At the age of 15 years old, she was diagnosed of multiple neurofibroma. At the age of 48 years old, she received the fibormectomy at a local hospital because of worsened symptoms of the left lower extremity. At day 1 after admission, the chest-enhanced CT revealed, a round soft tissue density sized 8.4 × 4.0 cm with unclear boundary with esophagus and heterogeneous enhancement in posteriormediastium, and multiple nodule-like soft tissue density shadows in subcutaneous chest and back (Fig. 2). Meglumine diatrizoate angiography of upper gastrointestinal tract revealed abnormal changes in the lower esophagus. Magnetic resonance imaging (MRI) revealed thoracic bone hyperplasia and disc deformation. Laboratory examination showed that the plasma level and urinary excretion of epinephrine, norepinephrine and dopamine were all in the normal range. The liver function and kidney function were normal.

Chest enhanced CT showing a round soft tissue density sized 8.4 × 4.0 cm (red arrow)
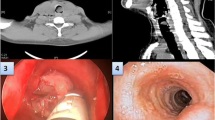
Before operation, the patient was treated with intravenous fluid resuscitation for a week and prepared for a surgery. A single-stage thoracoscopic mediastinal mass resection was performed. The patient was placed in the right supine position and anesthetized by double lumen tracheal intubation. A about 1.5 cm-long incision in the 9th intercostal axillary midline was made and thoracic exploration was performed by thoracoscope first. Then, a standard 24 cm-long lateral incision in the 7th intercostal space on the left was made. After each layer was cut, switch to single lung ventilation. No effusion or adhesions in the thoracic cavity and no space occupying lesion in pulmonary lobe were observed. The lower mediastinal pleural bulged and a mass adhered to surrounding tissue could be felt behind the esophagus and in front of the aorta. No obvious enlarged lymph nodes were found in the mediastinum. Mediastinal mass was diagnosed intraoperatively. Then, the posterior mediastinal pleura was open. After fully dissociating mass with surrounding tissues, the mass was completely resected. Rapid intraoperative pathology reported posterior mediastinal tumor. The tumor was measured about 11 × 7 × 5 cm in size with abundant blood supply and incomplete capsule (Fig. 3a). The tumor location was marked by titanium clip and a drainage tube was placed and fixed in the 9th intercostal space of the left axillary midline. Finally, suture the chest wall layer by layer. A total of 1000 mL fluid without transfusion was given. The whole operation went smoothly and the patient was back to ward safely.

Intraoperative image showing a tumor with abundant blood supply and incomplete capsule (a), and postoperative image in histopathological examination (b)
Postoperative histopathological examination showed the tumor was MPNST and measured 9.0 × 7.0 × 4.8 cm in size (Fig. 3b). Hematoxylin–eosin (H&E) staining showed obvious tumor cell atypia and irregular nuclei > 10/10 HPF (High power field) (Fig. 4). Immunohistochemistry revealed CD34(−), Desmin(−), Ki-67(+ 30%), SMA( +), Vimentin( +), S-100( +), CD99( +), Bcl-2(−), CD117(−), EMA( +), and Calretinin( +) (Fig. 5). Moreover, histopathological examination showed the mass in the body surface was benign neurofibroma and immunohistochemistry revealed Ki-67(+ 1%) and S-100( +). Three months later, the CT scan of lungs did not show relapse of tumor (Fig. 6).

Hematoxylin–eosin (H&E) staining showed obvious tumor cell atypia and irregular nuclei > 10/10 HPF (High power field). a 40×; b 100×; c 200×; d 400×

Immunohistochemistry staining of S-100 showing positive staining. a 100×; b 200×

The CT scan of lungs did not show relapse of tumor after 3 months
Discussion and conclusions
NF1 with an global prevalence of approximately 1 in 3000, is more common than NF2 with a birth incidence from 1 in 25,000 to 40,000 by estimate [1,2,3,4,5]. Patients with NF1 may have a higher risk of cancer, and a decrease in life expectancy of ~ 8 to 21 years, especially a higher mortality for those younger than 40 years old [2, 6, 7].
This inherited disease is strongly associated with gene Nf1. The result of Le et al. showed loss of Nf1 in skin-derived neural progenitors is required but not sufficient to induce tumors and suggested an essential role for the tumor microenvironment in neurofibroma development [8].
The clinical manifestations of NF1 include milk coffee spot, multiple neurofibroma, neurological symptoms, bone damage and visceral damage. As is known, neurofibromas are benign peripheral nerve sheath tumors arising from Schwann cell progenitors, of which cutaneous neurofibromas (cNF) are the most common type. The appearance of cNF varies, including nodular masses, peduncular lesions and diffuse plaques. They can be single or multiple, and are localized rather than encapsulated with no clear association with myelinated nerves [9]. Generally, cNF start developing early in adolescence, and keep increasing in number through the whole adulthood, which rarely transform into malignancy [10]. Surgical treatment is the preferred treatment for neurofibroma.
As for histology, S100 + Schwann cells were observed in surficial neurofibroma and part of posterior mediastinal MPNST. The significant difference between Ki67 expression in both tumor (1% of neurofibroma vs. 30% of nerve sheath tumor) indicated the dramatic cell proliferation. The positive biomarkers of nerve sheath tumor showed a possibility of its metastasis. Neurofibromas are mixed, consisting of neoplastic Schwann cells and non-neoplastic elements [8]. Usually, the diverse cellular components are embedded in collagenous and myxoid extracellular matrix [9]. Researches on finding new diagnostic tool with high sensitivity for MPNST never stop, such as Hirbe et al. reported β-III-spectrin immunohistochemistry as a potential diagnostic tool [11]. Besides traditional H&E staining and immunohistochemistry, a methylation-based classification of benign and malignant peripheral nerve sheath tumors was put forward, suggesting that the application of methylation status examination may facilitate the diagnosis of MPNST distinguish from benign nerve sheath tumors [12].
MPNST is a rare neoplasm of the peripheral nervous system. Most of the tumors occur in young and middle-aged men, and most of them occur in limbs, scalp and neck [13]. Atypical neurofibroma (ANF), with pathologically increased variable cellularity, cytological atypia and fascicular growth patterns, was regarded as precursor lesions for MPNST [13]. Other researchers also reported some cases of MPNST in the mediastinum [14,15,16]. Taken together, the appearance of neurofibroma should draw particular attention to the possibility of developing MPNST. Once NF1 is diagnosed, more careful imaging examinations should be carried out and needle biopsy is a more confirmative approach if possible.
Availability of data and materials
Not applicable.
Abbreviations
- NF1:
-
Neurofibromatosis type 1
- NF2:
-
Neurofibromatosis type 2
- MPNST:
-
Malignant peripheral nerve sheath tumor
- CT:
-
Computed tomography
- MRI:
-
Magnetic resonance imaging
References
Lin AL, Gutmann DH. Advances in the treatment of neurofibromatosis-associated tumours. Nat Rev Clin Oncol. 2013;10(11):616–24.
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. 2017;3:17004.
Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer: the neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15(5):290–301.
Rubin JB. Neurofibromatosis type 1—a model for nervous system tumour formation? Nat Rev Cancer. 2005;5(7):557–64.
Asthagiri ARPD, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, Lonser RR. Neurofibromatosis type 2. Lancet. 2009;373(9679):1974–86.
Uusitalo E, Leppavirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, Poyhonen M, Peltonen J, Peltonen S. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol. 2015;135(3):904–6.
Evans DG, O’Hara C, Wilding A, Ingham SL, Howard E, Dawson J, Moran A, Scott-Kitching V, Holt F, Huson SM. Mortality in neurofibromatosis 1: in North West England: an assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet EJHG. 2011;19(11):1187–91.
Le LQ, Shipman T, Burns DK, Parada LF. Cell of origin and microenvironment contribution for NF1-associated dermal neurofibromas. Cell Stem Cell. 2009;4(5):453–63.
Ortonne N, Wolkenstein P, Blakeley JO, Korf B, Plotkin SR, Riccardi VM, Miller DC, Huson S, Peltonen J, Rosenberg A, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91(2 Suppl 1):S5–13.
Jouhilahti EM, Peltonen S, Callens T, Jokinen E, Heape AM, Messiaen L, Peltonen J. The development of cutaneous neurofibromas. Am J Pathol. 2011;178(2):500–5.
Hirbe AC, Zhang X, Dahiya S, Godec A, Chrisinger J, Tao Y, Luo J, Gutmann DH. β-III-spectrin immunohistochemistry as a potential diagnostic tool with high sensitivity for malignant peripheral nerve sheath tumors. Neuro Oncol. 2018;20(6):858–60.
Röhrich M, Koelsche C, Schrimpf D, Capper D, Sahm F, Kratz A, Reuss J, Hovestadt V, Jones DTW, Bewerunge-Hudler M, et al. Methylation-based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016;131(6):877–87.
Higham CS, Dombi E, Rogiers A, Bhaumik S, Pans S, Connor SEJ, Miettinen M, Sciot R, Tirabosco R, Brems H, et al. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro Oncol. 2018;20(6):818–25.
Shimoyama T, Yoshiya K, Yamato Y, Koike T, Honma K. Long-term survival after removal of a malignant peripheral nerve sheath tumor originating in the anterior mediastinum. Gen Thorac Cardiovasc Surg. 2009;57(6):310–4.
Kawachi R, Takei H, Furuyashiki G, Koshi-Ishi Y, Goya T. A malignant peripheral nerve sheath tumor of the mediastinum in a patient with neurofibromatosis type 1: report of a case. Surg Today. 2008;38(10):945–7.
Bacha S, Chaouch N, Ayadi A, Zidi A, Cheikhrouhou S, Racil H, Chabbou A. Malignant peripheral sheath nerve tumor: an exceptional mass of the anterior and middle mediastinum. Rev Pneumol Clin. 2015;71(6):364–8.
Acknowledgements
Not applicable.
Funding
This study was supported by Jilin Provincial Department of science and technology (3D5195876428).
Author information
Authors and Affiliations
Contributions
YL designed this study. YZ, HC and GL collected the information and images. YZ, HC and GL wrote the manuscript. YL reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted under approval of the Ethics Committee of First Hospital of Jilin University.
Consent for publication
Written informed consent was obtained from the patient for publication of the case.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Cai, H., Lv, G. et al. A giant posterior mediastinal malignant peripheral nerve sheath tumor and benign neurofibroma in body surface: a case report. BMC Surg 21, 128 (2021). https://doi.org/10.1186/s12893-021-01122-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12893-021-01122-5