
Abstract
Background
In treating patients with cervical ossification of the posterior longitudinal ligament (COPLL), a novel surgery technique - anterior controllable antedisplacement and fusion (ACAF) suggested promising clinical benefits in recent exploratory studies.
Methods
This is a multicentre, randomized, open-label, parallel-group, active controlled trial that will compare the clinical benefits of ACAF versus conventional posterior laminoplasty (LAMP) in severe COPLL patients. A total of 164 patients will be enrolled and randomized in a 1:1 ratio to either ACAF or LAMP group. The primary efficacy measure is cervical- Japanese Orthopaedic Association (C-JOA) recovery rate at 12 months post operation, which is to be derived by Hirabayashi’s method from JOA data (range, 0 [worst] to 17 [normal condition]). Other important secondary efficacy endpoints include visual analogue scale (VAS) pain score (range, 0 [no pain] to 10 [most severe]), 10-item neck disability index (NDI, a total range of 0 to 50 points, the highest index the worst) and 6-level Nurick disability grade (range, 0 [mild] to 5 [severe]). Safety endpoints including adverse events, perioperative complications, and adverse events of special interest will also be assessed in this study. Full analysis set for baseline and efficacy data analyses according to the intention-to-treat principle will be established as the primary analysis population. Analysis of covariance (ANCOVA) will be used to analyze the C-JOA recovery rate, with random stratification factors (if appropriate) and the treatment group as fixed factors, and the baseline level of C-JOA score as covariate.
Discussion
This study is designed to demonstrate the clinical benefits of ACAF as compared to conventional LAMP in COPLL patients. It will provide clinical evidence that the novel surgery technique – ACAF might be more favorable in treating patients with severe cervical ossification of the posterior longitudinal ligament. (Words: 290).
Trial registration
ClinicalTrials.gov number, NCT04968028.
Similar content being viewed by others


Background
Ossification of the posterior longitudinal ligament (OPLL) is a pathologic process of lamellar bone deposition at the site of the posterior longitudinal ligament (PLL) [1]. And it often occurs in the cervical spine, which was first noticed in the Japanese patients and reported more commonly in Asian countries than North America, with a prevalence rate of 2 to 4% in Japan [2], 3.1% in China [3], 5.1% in India [4] and 2.5% in USA [5], respectively. As one kind of chronic and multifactorial disease, it usually causes spinal cord compression over time and lead to a reduced range of cervical motion [1],, and further serious neural injury like axial neck pain, diminished sensation, motor weakness, and changes in balance or gait stability, or more severely, incontinence, paralysis, severe respiratory failure and even death [1, 4,5,6].
Non-operative management can be often undertaken for asymptomatic patients without severe cord compression or myelomalacia on radiographic scans [6,7,8,9]. However, for patients with neurologic symptoms such as myelopathy or radiculopathy or evidence of spinal cord compression, surgical intervention is required and also the only effective treatment options to date [6,7,8,9,10]. Mainstream surgical approaches available currently are those utilizing anterior (ACCF, anterior cervical corpectomy and fusion), posterior (LAMP, laminoplasty; LF, laminectomy and fusion), or combined anteroposterior strategy, each with different levels of risks and benefits [1, 11]. Anterior approaches are technically more demanding and associated with higher rates of spinal cord injury, cerebrospinal fluid leak, dysphagia, dysarhtria, hoarseness but offer direct decompression for ventral OPLL pathology. Although posterior own higher safety, associated with higher rates of C5 palsy, axial pain, and may allow for ossification progression that lead to recurrence of neurological deterioration [11, 12]. Even so, high level of clinical evidence is still inconclusive so far [12], therefore, surgeons and patients have to subjectively decide on surgery strategy after individualizing the potential benefits of surgical decompression against the potential medical and surgical complications [1, 12].
To make full use of the estimated advantages of anterior and posterior approaches and maximally avoid disadvantages, we have designed a novel surgery technique - anterior controllable antidisplacement and fusion (ACAF) strategy in a hope to achieve anterior direct decompression without removing the OPLL [13,14,15]. Our preliminary results in the recent exploratory studies [13, 14], along with those found in a Korean case series report (N = 24) [16] suggested that ACAF technique is an effective and safe option for anterior decompression surgery in severe OPLL patients. Given a worldwide paucity of level-I evidence comparing surgical approaches to OPLL [11] and potential expert consensus or clinical practice guideline to come out, we designed this adequate and well-controlled investigation across multiple study sites in China. Here we present its rationale, methodology and analysis plan on how to compare the clinical effectiveness of ACAF technique against LAMP among severe COPLL patients. The randomized controlled trial aims to provide clinical evidence that ACAF might be more favorable in treating severe COPLL patients.
Methods and design
Study design
The STAR study (ClinicalTrials.gov number, NCT04968028) is a multicentre, randomized, open-label, parallel-group, active controlled trial that aims to demonstrate more favorable clinical benefits of ACAF as compared to conventional LAMP in severe COPLL patients. It will be conducted following the Consolidated Standards of Reporting Trials (CONSORT) statement (http://www.consort-statement.org/). Approximately twelve clinical centres in China will participate in this study. A brief flowchart of this study is provided in Fig. 1.

Study flow chart. Abbreviations: ACAF, anterior controllable antedisplacement and fusion; C-JOA, cervical Japanese Orthopaedic Association; LAMP, posterior laminoplasty; NDI, neck disability index; VAS, visual analogue scale
Study patients
A total of 164 eligible patients will be enrolled and randomized after screening at the study sites. The following are the inclusion criteria:
-
1.
The patient’s age is 18–70 years old, regardless of gender;
-
2.
The patient was diagnosed as severe ossification of the posterior longitudinal ligament (X-ray or computed tomography [CT] showed ossification of the posterior longitudinal ligament). The imaging findings showed occupied ratio ≥ 60% or involved three or more segments.
-
3.
Symptoms of spinal cord and nerve root compression, or accompanied by spinal cord compression symptoms such as dysfunction of urination and defecation, conservative treatment was ineffective or aggravated gradually;
-
4.
The participant (or his legal guardian) can sign the informed consent.
The exclusion criteria are as follows:
-
1.
Congenital malformations (including but not limited to occipitocervical malformations, congenital cervical fusion, cervical related neurovascular malformations), ossification of cervical ligamentum flavum, cervical trauma, cervical cancer, cervical tuberculosis and other inflammatory diseases;
-
2.
Patients with other spinal diseases such as thoracolumbar vertebrae that affect clinical symptoms; Patients with motor neuron diseases such as amyotrophic lateral sclerosis and other nervous system diseases;
-
3.
The symptoms were aggravated due to recent trauma;
-
4.
Patients who participated in other clinical trials in recent 3 months;
-
5.
The participant (or his legal guardian) with mental illness and cognitive impairment cannot give full informed consent;
-
6.
Those who are in poor health and cannot tolerate surgery; the patients were not suitable for surgical treatment after preoperative examination.
Recruitment and randomization process
Before enrollment, there will be one pretreatment screening visit at study site office, during which each participant will be assigned a unique identification number.
Once considered eligible for entry, these COPLL patients will be randomly assigned to one of two study treatment groups, e.g. either ACAF or LAMP surgery in a 1:1 ratio. A stratified randomization using minimization technique [17, 18] will be carried out, stratified by study site, occupation ratio (< 60% versus ≥60%), K-line status (negative versus positive) and count of vertebrae involved (< 3 versus ≥3). Random assignment is generated by an independent statistician and implemented via central randomization mobile phone APP (Shanghai KNOWLANDS MedPharm Consulting Co., Ltd.). In order to avoid potential selection bias, the randomization sequence is concealed in APP from both clinical staff and patients until assignment. Hence, neither any investigators nor patients can influence which treatment group the study patients are assigned to.
Description of the interventions
The enrolled subjects will be randomized to undergo the novel ACAF or conventional LAMP surgery. In order to minimize the potential bias of the trial data, all patients would be operated on by senior spine surgeons from spine surgery team at each site who are highly experienced in performing the two trial interventions. All patients in the study will accept the same surgical devices. All the consumables used in the operation are from the same manufacturer, and the operations will be carried out in strict accordance with the unified standard.
ACAF group
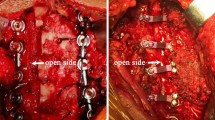
The surgical steps of ACAF were as follows: (1) a standard right-side Smith–Robinson approach was performed to expose the cervical spine, and intraoperative fluoroscopy was used to confirmed surgical levels. (2) The involved disc tissue was completely removed. The posterior longitudinal ligament was only cut down at the levels cephalic and caudal to OPLL, and the osteophytes at the anterior and posterior borders of vertebrae were appropriately removed for placing cages at the middle disc levels. (3) The anterior portion of the middle vertebral bodies with OPLL was appropriately removed according to the thickness of ossification. Suitable cages filled with autologous bone fragments were placed into each intervertebral space. (4) On the left side of the vertebra, high-speed burr and Kerrison rongeur were used to create a 2-mm-wide groove on the lateral side of vertebral body, approximately at the medial border of the transverse foramina. After that, an appropriate curved plated was fixed to the caudal and cephal ad vertebrae. On the middle vertebrae, screws were inserted halfway for temporary fixation. (5) On the right side of the vertebrae, as had performed on the left side, a similar groove was also created, and the vertebrae with OPLL were completely isolated. (6) Finally, the loose screws on the middle vertebrae were tightened to achieve a gradual evaluation of the vertebrae with OPLL, resulting in decompression of the spinal cord (Figs. 2A – 2E). The SKYLINE anterior cervical plate system with PEEK cage (Depuy Synthes Spine, Inc., Raynham, MA) was used in all patients. After surgery, all patients were immobilized in a Philadelphia collar for 6 to 8 weeks.

Schematic diagram of ACAF technique with steps A through E: (A) Expose and confirm surgical levels; (B) Remove discs, decompress at the levels cephalic and caudal to OPLL and remove the anterior portion of the middle vertebral bodies with OPLL; (C) Place interbody cages and create a groove on the left side, and fix anterior plate; (D) Create a groove on the right side, and isolate the vertebrae with OPLL and (E) Evaluate the vertebrae with OPLL. Sagittal (left) and transverse (right) sectional images of the cervical spine. Abbreviations: ACAF, anterior controllable antedisplacement and fusion; OPLL, ossification of the posterior longitudinal ligament
LAMP group
The surgical procedure of laminoplasty will be done as described in the literature using the ARCH miniplate fixation system (Depuy Synthes Spine, Inc., Raynham, MA). After surgery, all patients will be immobilized in a Philadelphia collar for 3 to 4 weeks.
As for the other treatments other than study surgery, including the perioperative treatment, study patients in the two groups both will follow Chinese Experts Consensus (2018) [19].
Study visits
Six study visits per subject are scheduled in the study as follows: pre-treatment visit (Day − 28 to Day − 1), perioperative visit (Day 1 until discharge) and follow-up visit at month 3 ± 1 week, month 6 ± 1 week, month 12 ± 2 week and month 24 ± 4 week (Month 3, 6, 12 and 24 post treatment). These visits will be made at study site office or patient ward as appropriate. At scheduled visits, data relating to demography, vital sign, operating time, estimated blood loss during operation period, length of stay at hospital in days, and perioperative complications, cervical Japanese Orthopaedic Association (C-JOA) score and visual analogue scale (VAS) pain score, Neck Disability Index (NDI), Nurick score, concomitant medication / treatment, new adverse events, radiographic data, revision surgery etc. will be collected. See Fig. 1 for more details.
In case severe adverse events occur, the subject can drop out at any time during the study.
Outcome measures
Primary efficacy outcome
The primary efficacy endpoint is the C-JOA recovery rate at 12 months post operation, which is to be derived by Hirabayashi’s method [20] from original C-JOA scores. A set of C-JOA score per subject visit involves four aspects: upper limb motor function (range, 0 to 4 points), lower limb motor function (range, 0 to 4 points), sensory function (range, 0 to 6 points), and bladder function (range, 0 to 3 points). The total C-JOA score ranged from 0 (worst) to 17 (normal condition), with a lower score indicating more significant dysfunction. The recovery rate of C-JOA is calculated by the following Hirabayashi’s formula [20]:
Secondary outcomes
The secondary efficacy outcomes in this study included VAS pain score (range, 0 [no pain] to 10 [most severe]) [21], 10-item NDI (a total range of 0 to 50 points, the highest index the worst) [22] and 6-level Nurick disability grade (range, 0 [mild] to 5 [severe]) [23].
Safety endpoints.
The safety endpoints include any adverse events (AEs), perioperative complications, and adverse events of special interest. The AEs profile of both treatments will be evaluated by examining the incidence of AEs according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE, Version5.0) [24].
Sample size calculation
We used SAS® software, V.9.4 (SAS Institute, North Carolina, USA) to estimate sample size. According to the principle of efficacy superiority design, the significance level α was set as 0.05 at two-sided. The primary efficacy endpoint of this study is the recovery rate of C-JOA at the 12th month of follow-up post operation. Assume that in severe COPLL patient population, the mean recovery rate of C-JOA in the ACAF group and the LAMP group at 12 months after operation is 63 and 51% (a mean difference of 12%), respectively, and the common standard deviation is 20%. When the two groups are randomly assigned 148 subjects in a 1:1 ratio, the statistical power can reach as high as 95%. If the subject dropout rate is 10% or less, 164 eligible subjects with severe COPLL should be randomly enrolled.
Statistical analysis
The study includes three analysis populations per analysis plan. For the baseline and efficacy data, we will use full analysis set (FAS), which includes all subjects who were randomized into the study groups and received the study operation scheme. According to the principle of intention to treat, subjects in FAS would be analyzed using their randomly assigned group, regardless of the actual surgical treatment received. Per-protocol population, e.g. a subset of FAS will be established further for subjects with no major protocol deviation(s), which will be reviewed and determined by the sponsor before the study database is locked. Safety set (SS) is defined to include all subjects who have received the treatment of the study-specified operation (regardless of whether they participate in the randomized assignment or not) and will be the primary analysis population for safety data. The subjects in SS will be grouped according to the actual surgical treatment received.
In this study, a two-sided p-value of 0.05 or less (equivalently one-sided p-value of 0.025) will be considered to indicate significance for any statistical tests, unless otherwise specified in the interim analysis section. Software R, V.4.0.4 [25] and SAS®, V.9.4 will be used for statistical analysis. Such data as demographics, baseline characteristics, surgery operation, radiographic data and safety will be summarized according to treatment group.
The primary endpoint of this study is the C-JOA recovery rate at 12 months post operation. It will be analyzed with the use of analysis of covariance (ANCOVA), with random stratification factors (if appropriate) and the treatment group as fixed factors, and the baseline level of C-JOA score as covariate. The least squares mean (LSM), the difference from the control group and the confidence interval (CI) of each treatment group will also provided for statistical analysis.
In addition, a mixed model repeated measures (MMRM) will be used as supportive analysis when appropriate. MMRM analysis includes treatment group, time and time treatment group interaction as fixed effects, baseline level as covariate, and patients as random effects. When there are missing data, multiple imputation method will be applied as the primary analysis; those results from the last observation carry forward method (LOCF) and the as-observed data analysis will play roles as sensitivity analyses. If the data distribution does not meet the statistical hypothesis of ANCOVA, a set of rank-based analysis will be the primary one. The LSM, the difference from the control group and the CI of each treatment group will be also provided for statistical analysis. For MMRM and ANCOVA, the two-sided p-values will be provided when compared with the control group.
To investigate the consistency of the trial conclusions among different subpopulations, the subgroup analysis with regards to the primary efficacy endpoint will include but not limited to the following baseline factors: gender; age group; duration of complaining symptoms; diabetes status; smoking status; count of vertebrae segments involved; type of COPLL; K-line status; occupation ratio; C-JOA score; Nurick grade and status of hypersignal of spinal cord. The occupation ratio (OR) is defined as the biggest ratio of COPLL thickness to anteroposterior diameter of the bony spinal canal on the axial computed tomography image. A forest plot will be also provided as a useful display of estimated treatment effects across subgroups.
For VAS pain score and NDI index as continuous variables, the analysis method of between-group comparison is similar to those used for the primary endpoint. For cervical Nurick grades as an ordinal variable, the common odds ratio will be estimated by using ordinal logistic regression analysis (proportional odds model) to reflect the shift of full distribution of each grade between the two groups 12 months after operation. The adjusted and unadjusted common odds ratio and its 95% CIs will be calculated. When using the model to adjust prognostic factors, the important prognostic factors mainly include random stratification factor as appropriate, gender, age and baseline Nurick score, which will be clearly specified in the statistical analysis plan before the database lock.
For purposes indicative of statistical significance, we will use the Cochran Mantel Haenszel (CMH) method according to random stratification factors as appropriate to test the statistical hypothesis for important categorical adverse events. The incidences in each group and 95% CIs of incidence difference between the two groups will be calculated by Newcombe method [26]. Similar statistical analysis will also be conducted for each single adverse event of special interest, or Fisher’s exact test will be used for comparison between groups when applicable.
Radiographic data based on central review as their primary analyses will be compared between groups. When appropriate, we will also conduct exploratory analysis on the relationship between these imaging data and clinical outcomes, with an effort to predict the long-term clinical outcomes and complications with the use of preoperative and recent postoperative imaging data as well as other subject-level baseline characteristics. The statistical methods used for these exploratory analyses will be depicted in more details in the statistical analysis plan as appropriate.
Interim analysis
Two formal interim analyses of efficacy data are planned when one-third and two-thirds of the enrolled subjects have completed at least 12 months of follow-up, respectively. The treatment efficacy and the occurrence of adverse events will be then reviewed by an independent Data and Safety Monitoring Board (DSMB), when the sample size might be also re-estimated and adjusted based on accumulative data of primary efficacy endpoint as necessary (Cui L et al., 1999) [27].
In the interim analysis of efficacy, the Haybitt-Peto boundary will be used (Haybitt 1971 [28]; Peto et al. 1976 [29]) to control the overall type I error. The alpha boundary is to be consumed at 0.0001 (one-sided and nominal level) at each interim analysis. The boundary value at the final stage will be kept at 0.025 at one-sided after the two scheduled interim analyses or be derived to maintain the Type I and Type II error probability levels if any ad hoc interim analysis is added.
Study committees
Four committees are planned in the whole study. A Steering Committee was well organized to oversee the governance of the study. From independent external oversight, a DSMB was established and chaired by a surgical expert and also included another DSMB statistician and one radiologist as well. The members of the DSMB serve in an individual capacity and provide their expertise and recommendations on a timely basis to the study Steering Committee.
A Technical Training Committee (TTC) was organized to develop a training manual on ACAF technique, carry out field lecture at each branch centre and perform demo surgery as appropriate. Besides, all of the radiographic scans (X-ray, CT and magnetic resonance imaging [MRI]) in the study will be forwarded to an independent Radiographic review Committee (IRC) for retrospective review on a regular basis.
Discussion
In the three recent exploratory studies [13, 14, 16], the pilot use of ACAF technique (or called vertebral body sliding osteotomy [VBSO] in principle) showed promising treatment benefits in COPLL patients. The novel surgery technique can achieve anterior direct decompression without separating and removing ossification. Its basic principle is to expand the spinal canal by anteriorly translating the involved vertebral bodies with any ossified masses [15, 16] (Fig. 2). Based on surgeons’ preference, this novel technique was initially proposed more suitable for severe COPPL patients as opposed to continue conventional anterior or posterior approaches for non-severe patients. Patients included in the small-sized studies are those with OPLL involved three or more vertebrae and a canal occupying ratio of more than 60% [13], or those with at least three levels of cervical compression caused by OPLL [14], or those with negative K-line and a large OPLL mass or kyphotic cervical alignment [16]. In line with these findings, we designed this new study to have a statistical hypothesis enriched in the severe COPLL subpopulation. To make a balanced assignment of ACAF versus LAMP across each stratum of patients and validity of subsequent statistical comparison, therefore, we pre-specified several stratification factors when conducting randomized assignment of the two treatments among enrolled subjects. This had better help set up any statistical modeling in the data analysis.
The use of anterior controllable antedisplacement and fusion belongs to one of anterior approaches, which are usually high technically demanding. However, the excellent neurologic outcome noted in the severe COPPL patients might be extended to the whole COPPL population as long as the spine surgeons in the study are rich in spine surgery experience and can also follow a standardized operation schema after uniform training held by TTC. This is why our study operation team in the sponsor’s site will design a training manual of ACAF technique, carry out field lecture and perform surgery in concert for the first several cases of each site. On the other hand, the study site is included as one of randomization factors, which provides a data basis for analyzing potential site effect in the future.
Furthermore, for purpose of better data quality, the study team will employ a dedicated clinical monitor team for source data verification. The radiographic data will be centrally reviewed by the IRC. Due to the limited OPLL patients at some of our study sites, it may take a long time to recruit the sample size required for this study. Moreover, we face the challenge of subjects’ follow-up issue to collect primary efficacy data especially in the long run. We will also utilize additional patient accrual and follow-up services from a dedicated third-party clinical research coordinator team. An electronic data capture system with audit trails will also be used in the study.
Based on this adequate and well-controlled study, we expect to obtain solid study data and with them to demonstrate whether this novel ACAF technique is more favorable than LAMP in the selected study population. We have a plan to formulate an expert consensus with our study findings and also develop a COPLL-specific clinical practice guideline as appropriate. All of these after publication will be shared with participating hospitals and the academic communities to promote the clinical management of severe COPLL patients.
Availability of data and materials
Not Applicable.
Abbreviations
- ACAF:
-
Anterior controllable antedisplacement and fusion
- ANCOVA:
-
Analysis of covariance
- C-JOA:
-
Cervical- Japanese Orthopaedic Association
- COPLL:
-
Cervical ossification of the posterior longitudinal ligament
- LAMP:
-
Posterior laminoplasty
- NDI:
-
Neck disability index
- VAS:
-
Visual analogue scale
- ACAF:
-
Anterior controllable antedisplacement and fusion
- ANCOVA:
-
Analysis of covariance
- CI:
-
Confidence interval
- C-JOA:
-
Cervical- Japanese Orthopaedic Association
- CMH:
-
Cochran Mantel Haenszel
- CONSORT:
-
Consolidated Standards of Reporting Trials
- COPLL:
-
Cervical ossification of the posterior longitudinal ligament
- CT:
-
Computed tomography
- DSMB:
-
Data and Safety Monitoring Board
- FAS:
-
Full analysis set
- IRC:
-
Independent Radiographic review Committee
- IEC:
-
Independent ethics committee
- LAMP:
-
Posterior laminoplasty
- LOCF:
-
Last observation carry forward
- LSM:
-
Least squares mean
- MMRM:
-
Mixed model repeated measures
- MRI:
-
Magnetic resonance imaging
- NCI CTCAE:
-
National Cancer Institute Common Terminology Criteria for Adverse Events
- NDI:
-
Neck disability index
- OPLL:
-
Ossification of the posterior longitudinal ligament
- SS:
-
Safety set
- TTC:
-
Technical Training Committee
- VAS:
-
Visual analogue scale
- VBSO:
-
Vertebral body sliding osteotomy
References
Barrett S. Boody, Mayan Lendner, Alexander R Vaccaro. Ossification of the posterior longitudinal ligament in the cervical spine: a review. Int Orthop. 2019;43(4):797–805.
Matsunaga S, Sakou T. OPLL: disease entity, incidence, literature search and prognosis. Tokyo: Springer; 2006. p. 11–7.
Zhen C, Sun Y. Research progress on epidemiology of ossification of posterior longitudinal ligament of cervical spine. Chin J Spine Spinal Cord. 2017;27(5):460–4.
Singh NA, Shetty AP, Jakkepally S, Kumarasamy D, Kanna RM, Rajasekaran S. Ossification of Posterior Longitudinal Ligament in Cervical Spine and Its Association With Ossified Lesions in the Whole Spine: A Cross-Sectional Study of 2500 CT Scans. Global Spine J. 2021;2192568221993440.
Bakhsh W, Saleh A, Yokogawa N, Gruber J, Rubery PT, Mesfin A. Cervical ossification of the posterior longitudinal ligament: a computed tomography-based epidemiological study of 2917 patients. Global Spine J. 2019;9(8):820–5. https://doi.org/10.1177/2192568219833658.
Nam DC, Lee HJ, Lee CJ, Hwang SC. Molecular pathophysiology of ossification of the posterior longitudinal ligament (OPLL). Biomol Ther (Seoul). 2019;27(4):342–8. https://doi.org/10.4062/biomolther.2019.043.
Pham MH, Attenello FJ, Lucas J, He S, Stapleton CJ, Hsieh PC. Conservative management of ossification of the posterior longitudinal ligament. A review. Neurosurg Focus. 2011;30(3):E2. https://doi.org/10.3171/2011.1.focus10273.
Matsunaga S, Sakou T. Ossification of the posterior longitudinal ligament of the cervical spine: etiology and natural history. Spine. 2012;37(5):E309–14. https://doi.org/10.1097/BRS.0b013e318241ad33.
Zhang J, Liang Q, Qin D, Song J, An Q, Wang X, et al. The anterior versus posterior approach for the treatment of ossification of the posterior longitudinal ligament in the cervical spine: a systematic review and meta-analysis. J Spinal Cord Med. 2019;6:1–10.
Aljuboori Z, Boakye M. The natural history of cervical Spondylotic myelopathy and ossification of the posterior longitudinal ligament: a review article. Cureus. 2019;11(7):e5074. https://doi.org/10.7759/cureus.5074.
Head J, Rymarczuk G, Stricsek G, Velagapudi L, Maulucci C, Hoelscher C, et al. Ossification of the posterior longitudinal ligament: surgical approaches and associated complications. Neurospine. 2019;16(3):517–29. https://doi.org/10.14245/ns.1938222.111.
Kwok SSS, Cheung JPY. Surgical decision-making for ossification of the posterior longitudinal ligament versus other types of degenerative cervical myelopathy: anterior versus posterior approaches. BMC Musculoskelet Disord. 2020;21(1):823. https://doi.org/10.1186/s12891-020-03830-0.
Sun J, Shi J, Xu X, Yang Y, Wang Y, Kong Q, et al. Anterior controllable antidisplacement and fusion surgery for the treatment of multilevel severe ossification of the posterior longitudinal ligament with myelopathy: preliminary clinical results of a novel technique. Eur Spine J. 2018;27(6):1469–78. https://doi.org/10.1007/s00586-017-5437-4.
Chen Y, Jingchuan S, Xiaoqiu Y, Guo Y, Yang H, Deyu C, et al. Comparison of anterior controllable Antedisplacement and fusion with posterior Laminoplasty in the treatment of multilevel cervical ossification of the posterior longitudinal ligament: a prospective, randomized, and control study with at least 1-year follow up. Spine. 2020;45(16):1091–101. https://doi.org/10.1097/BRS.0000000000003462.
Shi J. Principle and application of cervical spine anterior controllable antedisplacement and fusion. Beijing: Science Press; 2020.
Lee D-H, Park S, Hong CG. A novel anterior decompression technique for kyphosis line (K-line) ossification of posterior longitudinal ligament (OPLL): vertebral body sliding osteotomy. J Spine Surg. 2020;6(1):196–204. https://doi.org/10.21037/jss.2019.12.05.
Altman DG, Bland JM. Treatment allocation by minimisation. BMJ. 2005;330(7495):843. https://doi.org/10.1136/bmj.330.7495.843.
Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics. 1975;31(1):103–15. https://doi.org/10.2307/2529712.
Chinese Journal of Surgery. The experts consensus on the surgical treatment and perioperative management of cervical spondylosis. Chin J Surg. 2018;56(12):881–4. https://doi.org/10.3760/cma.j.issn.0529-5815.2018.12.001.
Hirabayashi K, Miyakawa J, Satomi K, et al. Operative results and postoperative progression of ossification among patients with ossification of cervical posterior longitudinal ligament. Spine. 1981;6(4):354–64. https://doi.org/10.1097/00007632-198107000-00005.
Gillian A, Hawker SM, Kendzerska T, French M. Measures of adult pain: Visual Analog Scale for Pain (VAS Pain), Numeric Rating Scale for Pain (NRS Pain), McGill Pain Questionnaire (MPQ), Short-Form McGill Pain Questionnaire (SF-MPQ), Chronic Pain Grade Scale (CPGS), Short Form-36 Bodily Pain Scale (SF-36 BPS), and Measure of Intermittent and Constant Osteoarthritis Pain (ICOAP). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S240–52.
Vernon H, Mior S. The neck disability index: a study of reliability and validity. J Manip Physiol Ther. 1991;14(7):409–15.
Nurick S. The pathogenesis of the spinal cord disorder associated with cervical spondylosis. Brain. 1972;95(1):87–100. https://doi.org/10.1093/brain/95.1.87.
National Cancer Institute (USA). Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 [http://evs.nci.nih.gov/ftp1/CTCAE/About.html, March 15, 2021, date last accessed].
R Core Team (2014). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. [URL http://www.R-project.org/, March 15th, 2021, date last accessed.]
Newcombe RG. Interval estimation for the difference between independent proportions: comparison of eleven methods. Stat Med. 1998;17(8):873–90. https://doi.org/10.1002/(SICI)1097-0258(19980430)17:8<873::AID-SIM779>3.0.CO;2-I.
Cui L, Hung HM, Wang SJ. Modification of sample size in group sequential clinical trials. Biometrics. 1999;55(3):853–7. https://doi.org/10.1111/j.0006-341X.1999.00853.x.
Haybittle JL. Repeated assessment of results in clinical trials of cancer treatment. Br J Radiol. 1971;44(526):793–7. https://doi.org/10.1259/0007-1285-44-526-793.
Peto R, Pike MC, Armitage P, Breslow NE, Cox DR, Howard SV, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient: I. Introduction and design. Br J Cancer. 1976;34(6):585–612. https://doi.org/10.1038/bjc.1976.220.
Acknowledgements
The authors wish to thank Yuhong Xiao (YX) for their generous assistance with this study.
Trial status
The study is not yet recruiting as of the date of manuscript submission.
Funding
This study was funded by Major Special Project of Three-Year Action Plan to Promote Clinical Skills and Clinical Innovation Ability of Municipal Hospitals, Shanghai Hospital Development Centre (Grant No. SHDC2020CR1024B). The funding body has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
JGS, YC and JCS conceived and designed the study. XHZ wrote the data analysis section. JGS bear overall responsibility for the design, ethical conduct and publication of the study. Administrative, technical and material support was provided by DH. All authors involved the protocol discussion and they will take responsibility for study data gathering and verification. All authors edited the draft and contributed substantially to the manuscript; they all approved this submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The independent ethics committee (IEC) of Changzheng Hospital approved the study protocol (Protocol code: CZ1812ACAF, Version 1.0, Issue date: 2021-06-21) for all of the currently planned participating centres (Approval No. 2021SL029). The IEC agreed that this study will not raise patients’ risk or cause extra harm to patients. The IEC further agreed that the study is in accordance with the Declaration of Helsinki. All subjects will be required to sign a written informed consent document before their participation in the study. The study had purchased medical insurance for any compensation to those who suffer harm from trial participation.
Consent for publication
Not Applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Sun, J., Han, D. et al. An open-label randomized multi-Centre study to evaluate anterior controllable Antedisplacement and fusion versus posterior Laminoplasty in patients with cervical ossification of the posterior longitudinal ligament: study design and analysis plan (STAR). BMC Musculoskelet Disord 22, 765 (2021). https://doi.org/10.1186/s12891-021-04645-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12891-021-04645-3