
Abstract
Background
Observational studies have shown that smoking is related to the diffusing capacity of the lungs for carbon monoxide (DLCO) in individuals with idiopathic pulmonary fibrosis (IPF). Nevertheless, further investigation is needed to determine the causal effect between these two variables. Therefore, we conducted a study to investigate the causal relationship between smoking and DLCO in IPF patients using two-sample Mendelian randomization (MR) analysis.
Methods
Large-scale genome-wide association study (GWAS) datasets from individuals of European descent were analysed. These datasets included published lifetime smoking index (LSI) data for 462,690 participants and DLCO data for 975 IPF patients. The inverse-variance weighting (IVW) method was the main method used in our analysis. Sensitivity analyses were performed by MR‒Egger regression, Cochran’s Q test, the leave-one-out test and the MR-PRESSO global test.
Results
A genetically predicted increase in LSI was associated with a decrease in DLCO in IPF patients [ORIVW = 0.54; 95% CI 0.32–0.93; P = 0.02].
Conclusions
Our study suggested that smoking is associated with a decrease in DLCO. Patients diagnosed with IPF should adopt an active and healthy lifestyle, especially by quitting smoking, which may be effective at slowing the progression of IPF.
Similar content being viewed by others

Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive interstitial lung disease (ILD) of unknown origin and has a poor prognosis [1]. Epidemiological studies reveal that the global incidence and prevalence of IPF are increasing annually, and the median survival duration following an IPF diagnosis is within the range of 3 to 5 years, with a five-year survival rate of under 30% [2, 3].
Cigarette smoking (CS) is a lifestyle factor that can potentially be modified, which consistently ranks as one of the primary risk factors for IPF. Therefore, CS have garnered significant attention as promising areas of intervention in efforts to prevent the risk of IPF and halt its progression [4]. However, research on the specific role of smoking in driving disease progression in individuals with IPF is still limited. diffusing capacity of the lung for carbon monoxide (DLCO) is an important indicator used to assess disease progression in patients with IPF and reflects the ability of the lungs of IPF patients to transfer gas [5, 6]. Previous observational studies have shown that both current smoking status and increasing pack-years of CS were linked to lower DLCO. This implies that individuals with a history of CS, particularly those who have smoked for an extended period or at a higher intensity, are more susceptible to a decline in DLCO [7,8,9,10]. In summary, smoking appears to be a contributing factor to decreased DLCO in patients diagnosed with IPF. However, the determination of smoking as a causal factor of decreased DLCO remains uncertain, given that these studies are primarily observational. These studies are susceptible to confounding bias and reverse causality, which can complicate the interpretation of the relationship between smoking and DLCO in patients with IPF.
Mendelian randomization (MR) is a new approach that addresses these above challenges by using genetic variants as a reliable tool for establishing causal relationships; this approach is less vulnerable to confounding bias than is conventional observational studies [11]. In this context, we conducted an MR study to explore the potential causal link between smoking and a decrease in DLCO.
In summary, our research aimed to investigate the causal relationship between smoking and DLCO in individuals with IPF via a two-sample MR approach. The research based on summary data of large-scale genome-wide association study (GWAS).
Methods
Study design
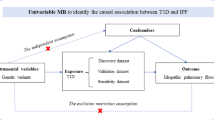
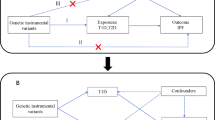
To evaluate the causal link between smoking and DLCO, we executed a two-sample MR analysis. The reliability of instrumental variables (IVs) hinges upon the fulfillment of three fundamental assumptions [12]. First, the genetic variants employed as IVs must be significantly associated with the targeted exposure. Second, these IVs should not be linked to any confounders. Finally, these IVs affect the outcome via alternative pathways (Fig. 1).

Workflow and the three assumptions for Mendelian randomization analysis
GWAS data sources
The primary metric for quantifying smoking behaviour was the lifetime smoking index (LSI), which was ascertained through a GWAS carried out in the UK Biobank; this study included 462,690 individuals of European descent, as reported by Wootton et al. [13]. The construction of the LSI involved the utilization of self-report questionnaire data regarding smoking intensity, duration, and initiation, following the methodology outlined by Leffondre et al [14]. This approach aimed to provide a more comprehensive representation of smoking habits. The study identified 124 genetic markers associated with the LSI, all of which reached genome-wide significance (P < 5 × 10− 8) and exhibited minimal linkage disequilibrium (LD) (r2 < 0.001).
The GWAS summary data for DLCO were derived from The Collaborative Group of Genetic Studies of IPF [15]. This comprehensive analysis of 3 cohorts (US, UK, and UUS) included the genotype data of 975 individuals diagnosed with IPF. The patients were diagnosed in accordance with the guidelines established by the American Thoracic Society and the European Respiratory Society.
Selection of genetic instruments
To ensure the reliability of the IVs used for MR analyses, we adhered to the following criteria. First, we chose single nucleotide polymorphisms (SNPs) associated with smoking-related traits, and the threshold value was P < 5 × 10− 8. Second, to prevent any LD among all IVs for IPF, we set the clumping parameter to R2 < 0.001 and a window size of 10 Mb. Third, during the harmonization process, we removed palindromic SNPs from the IV. Fourth, to mitigate the risk of bias stemming from weak IVs, we calculated the F-statistic \( (F={beta}^{2}/{se}^{2})\) [16]. If the F-statistic for IVs greatly exceeded 10, the likelihood of bias from weak IVs was minimal [17]. Moreover, all GWAS data utilized in our MR analyses were limited to individuals of European descent to exclude potential biases from population heterogeneity.
Mendelian randomization analyses
In our MR analysis, we chose the inverse variance weighted (IVW) method, which combines the Wald ratio for each SNP, as the primary approach, leading to a consolidated causal estimate [18, 19]. To ensure the robustness of our analysis and account for potential pleiotropy, we also conducted sensitivity analyses using several complementary methods. These methods included the weighted mode [20], MR‒Egger [21], weighted median [22], simple mode [23], and MR-Pleiotropy RESidual Sum and Outlier (MR-PRESSO) [24].
Sensitivity analysis
To identify potential pleiotropy and assess the robustness of our results, we conducted several analyses, including Cochran’s Q statistic [25] and MR‒Egger intercept tests [21]. Specifically, heterogeneity was indicated if the P value of the Cochran Q test was less than 0.05. We also assessed horizontal pleiotropy based on the intercept term derived from MR‒Egger regression. In addition, to ascertain whether any single SNP drove the causal estimate, we performed leave-one-out analysis [26].
MR analysis was performed using RStudio (version 4.2.1) with the TwoSampleMR (version 0.5.6) and MRPRESSO (version 1.0) R packages. A significance level of P < 0.05 was used to determine statistical significance.
Ethics
Summary data were used, and ethical approval was not needed.
Results
MR estimate
We identified 119 SNPs (Additional file 1) as IVs to investigate the genetic relationship between LSI and DLCO. The F-statistic for each SNP exceeding 30 indicated a low probability of a weak IV. Subsequently, we conducted an MR analysis utilizing these 119 SNPs. The results obtained through the IVW method revealed a causal link between the LSI and DLCO (ORIVW = 0.54, 95% CI 0.32–0.93; P = 0.02; Fig. 2). Furthermore, (ORMR−Egger = 0.09, 95% CI 0.01–0.73, P = 0.03, Fig. 2; (ORWeighted median = 0.41, 95% CI 0.18–0.90, P = 0.03, Fig. 2); (ORSimple mode = 0.23, 95% CI 0.03–2.02, P = 0.19, Fig. 2); (ORWeighted mode = 0.25, 95% CI 0.05–1.26, P = 0.1, Fig. 2); and (ORMR−PRESSO = 0.54, 95% CI 0.32–0.93, P = 0.03, Fig. 2) had consistent directions of effects across all six methods. As illustrated in the scatter plot (Fig. 3A), there was a noticeable decrease in DLCO as the LSI increased.

Causal effect of the lifetime smoking index on DLCO
Sensitivity analyses
We subsequently conducted sensitivity analyses to assess the robustness of our results. First, Cochran’s Q test demonstrated the absence of heterogeneity among the IVs (PMR−Egger = 0.374, PIVW = 0.325; Table 1). The absence of heterogeneity was also confirmed by the symmetry of the funnel plot (Fig. 3B). Second, there was no indication of overall horizontal pleiotropy across all IVs, as evidenced by the results of both the MR‒Egger regression (P = 0.085, Table 1) and the MR-PRESSO global test (P = 0.329, Table 1). These results imply that IVs are unlikely to exert their influence on the decrease in DLCO through pathways unrelated to smoking. In the leave-one-out sensitivity analysis, where we systematically excluded one SNP at a time, the results revealed that no specific SNP exerted a significant influence on the DLCO (Additional file 2). As a result, our results remained robust and exhibited no substantial bias.

Scatter plot (A) of the effect of the lifetime smoking index on DLCO and funnel plot (B)
Discussion
IPF patients have a median survival of 3–5 years after diagnosis but a highly variable clinical course [27]. Lung function in patients with IPF may decline precipitously from the onset of the disease or slowly over the course of the disease, during which acute exacerbations (AEs) occur that can lead to respiratory failure and early death [28]. Therefore, further research into the factors associated with the progression of IPF has become essential, as these factors can enhance the prevention of this condition and decelerate the progression of IPF.
Pulmonary function tests are essential for detecting, diagnosing, and monitoring the progression of IPF. However, given the infancy of computed tomography biomarkers, estimates of disease severity and risk stratification in IPF are still based almost exclusively on functional and physiologic indices, such as forceful lung volume (FVC), diffusing capacity for carbon monoxide (DLCO), and the 6-minute walk test (6MWT), with DLCO considered one of the most valuable parameters for monitoring the progression of IPF. DLCO is considered one of the most valuable pulmonary function test parameters for monitoring the progression of IPF [29,30,31].. To the best of our knowledge, this is the first study to determine the causal links between smoking and DLCO in patients with IPF based on the MR framework. Our approach drew upon large-scale GWAS data, allowing us to analyse a substantially larger number of cases than did previous observational studies. As expected, our study showed that smoking leads to negative effects on DLCO, which is largely in line with the findings of previous research [7,8,9,10]. While the link between smoking and IPF has been established in previous observational studies, our MR analysis offers robust evidence that aligns with the possibility of a causal connection, which is less vulnerable to confounding bias. Nevertheless, because we utilized summary-level data, we were unable to delve into sex-specific associations, indicating the need for future investigations in this area.
The specific mechanisms through which smoking exacerbates a decrease in DLCO have not been identified. Chronic lung inflammation and oxidative stress may be potential pathways mediating the relationship between smoking and reduced DLCO levels. Smoking harms the lungs by inciting chronic inflammation and oxidative stress, thus worsening the progression of IPF [32,33,34,35]. It leads to persistent inflammation, disrupts the balance of oxidation, contributes to the buildup of extracellular matrix in the lungs, impairs lung function, hampers gas exchange, and accelerates the deterioration of IPF [32, 36]. Exposure to CS or its extract (CSE) results in the senescence of alveolar epithelial type 2 (AT2) cells, a pivotal process in the progression of lung fibrosis [37]. Several mechanisms drive the CS-induced senescence of AT2 cells, including decreased autophagy, deactivation of the SIRT1 protein, DNA damage, and heightened oxidative stress. In addition, there is growing evidence of a potential correlation between smoking and a variety of IPF prognostic factors (such as MMP-7, SP-A, SP-D, GDF15, and CA-125). For example, higher levels of LOXL2 are associated with poor progression in IPF patients, and there is evidence that LOXL2 is significantly upregulated in patients who smoke. Moreover, SP-D, a serum marker, was found to be higher in smoking patients compared to non-smoking patients [31, 38, 39]. All these imply a potential relationship between CS and multiple IPF prognostic factors. Overall, CS is pivotal in additional damage to the lungs [40]. Concerning its public health implications, our discoveries lend support to the notion that smoking cessation initiatives can serve as an efficacious strategy for mitigating the decrease in DLCO and the ensuing adverse consequences.
Our study offers several notable advantages. First, an inaugural MR investigation was performed to evaluate the causal relationship between elevated smoking and decreased IPF. Second, the robustness of the analysis results was ensured by various sensitivity analysis methods. The study’s limitations must be acknowledged. First, our findings predominantly pertain to participants of European ancestry, and their applicability to populations of different racial backgrounds may be limited. Second, despite the absence of horizontal pleiotropy in our analysis, there may be residual bias due to limited knowledge about the precise functions of most of these SNPs. Third, as our study relied on GWAS summary data instead of individual-level data, it was not possible to stratify our analysis based on other variables, such as age and sex.
Conclusion
Our study suggested that smoking is an important factor for DLCO decline in IPF patients, which may provide new insights into the progression of IPF. Considering the imperative of delaying disease progression, significant emphasis should be placed on lifestyle management, including smoking cessation as a relevant strategy.
Data availability
All the data generated or analysed during this study are included in this published article and its Additional files.
Abbreviations
- AEs:
-
Acute Exacerbations
- AT2:
-
Alveolar epithelial type 2
- CS:
-
Cigarette smoking
- CSE:
-
Cigarette smoking extract
- DLCO:
-
Diffusing capacity of the lungs for carbon monoxide
- GWAS:
-
Genome-wide association study
- ILD:
-
Interstitial Lung Disease
- IPF:
-
Idiopathic pulmonary fibrosis
- IVs:
-
Instrumental variables
- IVW:
-
Inverse variance weighted
- LD:
-
Linkage disequilibrium
- LSI:
-
Lifetime smoking index
- MR-PRESSO:
-
MR pleiotropy residual sum and outlier
- MR:
-
Mendelian randomization
- SNPs:
-
Single nucleotide polymorphisms
References
Sgalla G, Biffi A, Richeldi L. Idiopathic pulmonary fibrosis: diagnosis, epidemiology and natural history. Respirology. 2016;21(3):427–37. https://doi.org/10.1111/resp.12683
Wu JSD, Li Z, et al. Immunity-and-matrix-regulatory cells derived from human embryonic stem cells safely and effectively treat mouse lung injury and fibrosis. Cell Res. 2020;30(9):794–809. https://doi.org/10.1038/s41422-020-0354-1
Liu P, Miao K, Zhang L, et al. Curdione ameliorates bleomycin-induced pulmonary fibrosis by repressing TGF-β-induced fibroblast to myofibroblast differentiation. Respir Res. 2020;21(1):58. https://doi.org/10.1186/s12931-020-1300-y
Gandhi S, Tonelli R, Murray M, Samarelli AV, Spagnolo P. Environmental causes of idiopathic pulmonary fibrosis. Int J Mol Sci. 2023;24(22):16481. Published 2023 Nov 18. https://doi.org/10.3390/ijms242216481
Kim JS, Ma SF, Ma JZ, et al. Associations of plasma Omega-3 fatty acids with progression and survival in pulmonary fibrosis. Chest Published Online Oct. 2023;20. https://doi.org/10.1016/j.chest.2023.09.035
Modi P, Cascella M. Diffusing capacity of the lungs for carbon monoxide. In: StatPearls. Treasure Island (FL): StatPearls Publishing; March 13, 2023.
Schwartz DA, Van Fossen DS, Davis CS, et al. Determinants of progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1994;149(2 Pt 1):444–9. https://doi.org/10.1164/ajrccm.149.2.8306043
Cherniack RM, Colby TV, Flint A, et al. Correlation of structure and function in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1995;151(4):1180–8. https://doi.org/10.1164/ajrccm/151.4.1180
Schwartz DA, Merchant RK, Helmers RA, Gilbert SR, Dayton CS, Hunninghake GW. The influence of cigarette smoking on lung function in patients with idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1991;144(3 Pt 1):504–6. https://doi.org/10.1164/ajrccm/144.3_Pt_1.504
Kreuter M. IPF, comorbidities and management implications: patient case 1. Sarcoidosis Vasc Diffuse Lung Dis. 2015;32(Suppl 1):24–5. Published 2015 Aug 3.
Richmond RC, Davey Smith G. Mendelian randomization: concepts and scope. Cold Spring Harb Perspect Med. 2022;12(1):a040501. https://doi.org/10.1101/cshperspect.a040501. Published 2022 Jan 4.
Lawlor DA, Harbord RM, Sterne JA, Timpson N, Davey Smith G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat Med. 2008;27(8):1133–63. https://doi.org/10.1002/sim.3034
Wootton RE, Richmond RC, Stuijfzand BG, et al. Evidence for causal effects of lifetime smoking on risk for depression and schizophrenia: a Mendelian randomisation study. Psychol Med. 2020;50(14):2435–43. https://doi.org/10.1017/S0033291719002678
Leffondré K, Abrahamowicz M, Xiao Y, Siemiatycki J. Modelling smoking history using a comprehensive smoking index: application to lung cancer. Stat Med. 2006;25(24):4132–46. https://doi.org/10.1002/sim.2680
Allen RJ, Oldham JM, Jenkins DA, et al. Longitudinal lung function and gas transfer in individuals with idiopathic pulmonary fibrosis: a genome-wide association study. Lancet Respir Med. 2023;11(1):65–73. https://doi.org/10.1016/S2213-2600(22)00251-X
Lei W, Yang M, Yuan Z, et al. The causal relationship between physical activity, sedentary time and idiopathic pulmonary fibrosis risk: a Mendelian randomization study. Respir Res. 2023;24(1):291. https://doi.org/10.1186/s12931-023-02610-3. Published 2023 Nov 20.
Burgess S, Thompson SG, CRP CHD Genetics Collaboration. Avoiding bias from weak instruments in Mendelian randomization studies. Int J Epidemiol. 2011;40(3):755–64. https://doi.org/10.1093/ije/dyr036
Lin T, Zhou F, Mao H, Xie Z, Jin Y. Vitamin D and idiopathic pulmonary fibrosis: a two-sample Mendelian randomization study. BMC Pulm Med. 2023;23(1):309. https://doi.org/10.1186/s12890-023-02589-z. Published 2023 Aug 23.
Burgess S, Davey Smith G, Davies NM, et al. Guidelines for performing Mendelian randomization investigations: update for summer 2023. Wellcome Open Res. 2023;4:186. https://doi.org/10.12688/wellcomeopenres.15555.3. Published 2023 Aug 4.
Hartwig FP, Davey Smith G, Bowden J. Robust inference in summary data Mendelian randomization via the zero modal pleiotropy assumption. Int J Epidemiol. 2017;46(6):1985–98. https://doi.org/10.1093/ije/dyx102
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–25. https://doi.org/10.1093/ije/dyv080
Bowden J, Davey Smith G, Haycock PC, Burgess S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40(4):304–14. https://doi.org/10.1002/gepi.21965
Chen Y, Zhao M, Ji K, et al. Association of nicotine dependence and gut microbiota: a bidirectional two-sample Mendelian randomization study. Front Immunol. 2023;14:1244272. https://doi.org/10.3389/fimmu.2023.1244272. Published 2023 Nov 7.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases [published correction appears in Nat Genet. 2018;50(8):1196]. Nat Genet. 2018;50(5):693–698. https://doi.org/10.1038/s41588-018-0099-7
Hemani G, Bowden J, Davey Smith G. Evaluating the potential role of pleiotropy in Mendelian randomization studies. Hum Mol Genet. 2018;27(R2):R195–208. https://doi.org/10.1093/hmg/ddy163
Burgess S, Bowden J, Fall T, Ingelsson E, Thompson SG. Sensitivity analyses for robust causal inference from Mendelian randomization analyses with multiple genetic variants. Epidemiology. 2017;28(1):30–42. https://doi.org/10.1097/EDE.0000000000000559
Harari S, Caminati A. IPF: new insight on pathogenesis and treatment. Allergy. 2010;65(5):537–53. https://doi.org/10.1111/j.1398-9995.2009.02305.x
Martinez FJ, Safrin S, Weycker D, et al. The clinical course of patients with idiopathic pulmonary fibrosis. Ann Intern Med. 2005;142(12 Pt 1):963–7. https://doi.org/10.7326/0003-4819-142-12_part_1-200506210-00005
Martinez FJ, Flaherty K. Pulmonary function testing in idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006;3(4):315–21. https://doi.org/10.1513/pats.200602-022TK
Capaccione KM, Wang A, Lee SM, et al. Quantifying normal lung in pulmonary fibrosis: CT analysis and correlation with %DLCO. Clin Imaging. 2021;77:287–90. https://doi.org/10.1016/j.clinimag.2021.06.021
Karampitsakos T, Juan-Guardela BM, Tzouvelekis A, et al. Precision medicine advances in idiopathic pulmonary fibrosis. EBioMedicine. 2023;95:104766. https://doi.org/10.1016/j.ebiom.2023.104766
Makena P, Kikalova T, Prasad GL, Baxter SA. Oxidative stress and lung fibrosis: towards an adverse outcome pathway. Int J Mol Sci. 2023;24(15):12490. Published 2023 Aug 6. https://doi.org/10.3390/ijms241512490
Pan M, Zheng Z, Chen Y, et al. Angiotensin-(1–7) attenuated cigarette smoking-related pulmonary fibrosis via improving the impaired autophagy caused by Nicotinamide Adenine Dinucleotide Phosphate Reduced Oxidase 4-Dependent reactive oxygen species. Am J Respir Cell Mol Biol. 2018;59(3):306–19. https://doi.org/10.1165/rcmb.2017-0284OC
Liu Y, Lu L, Yang H, et al. Dysregulation of immunity by cigarette smoking promotes inflammation and cancer: a review. Environ Pollut. 2023;339:122730. https://doi.org/10.1016/j.envpol.2023.122730
Lugg ST, Scott A, Parekh D, Naidu B, Thickett DR. Cigarette smoke exposure and alveolar macrophages: mechanisms for lung disease. Thorax. 2022;77(1):94–101. https://doi.org/10.1136/thoraxjnl-2020-216296
Obernolte H, Niehof M, Braubach P, et al. Cigarette smoke alters inflammatory genes and the extracellular matrix - investigations on viable sections of peripheral human lungs. Cell Tissue Res. 2022;387(2):249–60. https://doi.org/10.1007/s00441-021-03553-1
Zhang Y, Huang W, Zheng Z, et al. Cigarette smoke-inactivated SIRT1 promotes autophagy-dependent senescence of alveolar epithelial type 2 cells to induce pulmonary fibrosis. Free Radic Biol Med. 2021;166:116–27. https://doi.org/10.1016/j.freeradbiomed.2021.02.013
Chien JW, Richards TJ, Gibson KF, et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J. 2014;43(5):1430–8. https://doi.org/10.1183/09031936.00141013
Maher TM, Oballa E, Simpson JK, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. 2017;5(12):946–55. https://doi.org/10.1016/S2213-2600(17)30430-7
Douglas D, Keating L, Strykowski R, et al. Tobacco smoking is associated with combined pulmonary fibrosis and emphysema and worse outcomes in interstitial lung disease. Am J Physiol Lung Cell Mol Physiol. 2023;325(2):L233–43. https://doi.org/10.1152/ajplung.00083.2023
Acknowledgements
This study would not be possible without the summary GWAS data shared by Dr. Wootton RE and Dr. Allen RJ.
Funding
This study was supported by the Basic Research Program of Science and Technology Project of Yunnan Science and Technology Department [202101AY070001-286 to Mei Yang], Basic Research Program of Science and Technology Project of Yunnan Science and Technology Department [202101AY070001-285 to Jiyang Wang], and Famous Doctors of High-level Talent Training Support Program of Yunnan Province [No. YNWR-MY-2020-013].
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: ZHY, JYW and MY. Performed and analyzed the experiments: ZHY, JYW and WYL. Collected the data: SYQ and ZYY. Wrote the manuscript: XHH, XXH, and YYL. Improved the English writing: XQX and LW. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Additional Table.
Instrument variables of lifetime smoking index
Supplementary Material 2: Additional Figure.
Forest plot (A) and leave-one-out analysis (B) for lifetime smoking index on DLCO
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yuan, Z., Lei, W., Xing, X. et al. Genetic association between smoking and DLCO in idiopathic pulmonary fibrosis patients. BMC Pulm Med 24, 163 (2024). https://doi.org/10.1186/s12890-024-02974-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-024-02974-2