
Abstract
Background
Pulmonary arterial hypertension (PAH) is characterized by a progressive increase in pulmonary vascular resistance and pulmonary arterial pressure, with complex etiology, difficult treatment and poor prognosis. The objective of this study was to investigate the potential biomarkers for PAH based on bioinformatics analysis.
Methods
The GSE117261 datasets were downloaded from the Gene Expression Omnibus database. Differentially expressed genes (DEGs) were identified by screening PAH patients and controls. Then the DEGs were analyzed using a Weighted Gene Co-expression Network Analysis (WGCNA) and the key modules were determined, and to further explore their potential biological functions via Gene Ontology analysis (GO), Kyoto Encyclopedia of Genes and Genomes Pathway analysis (KEGG), and Gene Set Enrichment Analysis (GSEA). Moreover, Protein–protein interaction (PPI) networks were constructed to identify hub gene candidates in the key modules. Finally, real-time quantitative polymerase chain reaction was supplied to detect the expressions of hub genes in human pulmonary arterial smooth cells treated with cobalt chloride (COCl2) which was used to mimic hypoxia.
Results
There were 2299 DEGs identified. WGCNA indicated that yellow module was the key one correlated with PAH. GO and KEGG analysis demonstrated that genes in the yellow module were mainly enriched in ‘Pathways in cancer’. GSEA revealed that ‘HALLMARK_MYC_TARGETS_V1’ was remarkably enriched in PAH. Based on the PPI network, vascular endothelial growth factor A, proto-oncogene receptor tyrosine kinase (KIT), PNN interacting serine and arginine rich protein (PNISR) and heterogeneous nuclear ribonucleoprotein H1 (HNRNPH1) were identified as the hub genes. Additionally, the PCR indicated that the elevated expressions of PNISR and HNRNPH1 were in line with the bioinformatics analysis. ROC analysis determined that PNISR and HNRNPH1 may be potential biomarkers to provide better diagnosis of PAH.
Conclusion
PNISR and HNRNPH1 were potential biomarkers to diagnosis PAH. In summary, the identified DEGs, modules, pathways, and hub genes provide clues and shed light on the potential molecular mechanisms of PAH.
Similar content being viewed by others


Introduction
Pulmonary arterial hypertension (PAH) is a kind of severe cardiovascular disease, characterized by the progressive increase in pulmonary vascular resistance and pulmonary arterial pressure, with complex etiology and poor prognosis, finally leading to right heart failure and death [1]. PAH is the term used to describe the finding of a mPAP ≥ 25 mmHg regardless of the underlying cause [2]. At present, the treatment strategies of PAH mainly include supportive therapy such as diuretics and oxygen therapy, medication therapies and surgeries such as heart and lung transplantation [3]. Regrettably, there is still no effective treatment for PAH, and the mortality remains high [4]. In recent years, studies of gene expression chips have been widely used to find potential biomarkers in complex diseases in order to further explore underlying pathogenesis and potential therapeutic targets [5].
A weighted gene co-expression network analysis (WGCNA) is used to identify correlations between genes and microarray samples [6] and the association of genes and clinical traits could recognize by clustering genes with similar expression profiles. Therefore, diseases-related hub genes could be discovered via WGCNA in cardiovascular diseases [7] and cancers [8].
In this study, the gene expression profiles of GSE117261 from 32 PAH patients and 25 controls were re-analyzed. The “limma” package in R software was used to obtain the DEGs. WGCNA was conducted to build a gene co-expression network. GO and KEGG enrichment analysis were performed in the key module and hub genes in the key module were determined and validated by Real-time quantitative polymerase chain reaction (RT-qPCR).
Materials and methods
Data Sources
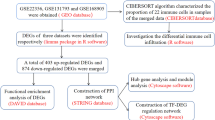
Microarray expression data (GSE117261) [9, 10] was downloaded from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/), representing a GPL6244 Affymetrix Human Gene 1.0 ST Array. GSE117261 includes 57 lung tissue specimens from 32 PAH patients and 25 normal controls. A workflow of the present study is presented in Fig. 1.

Workflow chart of the present study
Identification of differentially expressed genes
Data quality checking and normalization with log transformation were first performed to eliminate any batches. The “limma” package [11] in R software was used to screen DEGs between the PAH and control group. An adjusted P-value < 0.05 was set as the threshold criterion for statistical significance. The volcano map of DEGs was plotted in R software. The Heatmap package in R software was utilized to visualize the top 50 DEGs.
Weighted gene co-expression network analysis
WGCNA [12] was conducted to build the co-expression network in DEGs based on the scale-free topology criteria. First, all DEGs were analyzed using the WGCNA package in R software, and the soft thresholding power was set. Next, the weighted co-expression network was constructed, and DEGs were clustered into several modules with different color labels. The correlation between each module and PAH or controls was then explored. The module most correlated with PAH was regarded as a key module for further enrichment analysis.
Functional enrichment analysis of genes in the key module
Key module indicating the deep correlations with PAH were taken for further analysis. Base on the “org.Hs.eg.db” package and “cluster profiler” R package were installed in R software, GO and KEGG enrichment analysis was used to examine the gene complement of the key module [13,14,15,16,17,18]. The GO analysis annotates gene function contains: biological process (BP), cellular component (CC), and molecular function (MF). A P-value < 0.05 was considered statistically significant. Then bubble charts were used to display the top 10 enriched items from each GO category and from KEGG.
Gene set enrichment analysis
Gene set enrichment analysis (GSEA) [19] was performed using the hallmark gene sets (h.all.v7.0.symbols), which were obtained from the Molecular Signatures Database (https://www.gsea-msigdb.orggsea/msigdb/index.jsp), and the analysis was used to detect whether the relevant biological pathways is statistically significant in PAH patients and controls. Transcriptome data were imported into the GSEA software v4.0.3. The cutoff point of significance was normalized enrichment score |NES|> 1, P-value < 0.05, false discovery rate (FDR) q-value < 0.25 for GSEA.
Protein–protein interaction network and hub gene identification
The Search Tool for the Retrieval of Interacting Genes/Proteins (STRING, http://string-db.org/) was used to construct the protein–protein interaction (PPI) network of the key modules [20]. The PPI network was then visualized by Cytoscape software [21]. The Molecular Complex Detection (MCODE) [22] was used to screen the significant modules of the PPI network and then Cytohubba [23]was used to determine the hub genes.
Isolation and culture of human PASMCs
Human samples were collected with a protocol approved by the Branch for Medical Research and Clinical Technology Application, Ethics Committee of the First Affiliated Hospital of Fujian Medical University (Approval No. MRCTA, ECFAH of FMU[2021]483). The clinical samples of human pulmonary arterioles were obtained from 10 male patients (60 ± 11 years old) who underwent lobectomy for lung cancer in the Department of Thoracic Surgery, The First Affiliated Hospital of Fujian Medical University, Fujian Province. The normal pulmonary arterioles of patients undergoing clinical lobectomy were taken, and the blood vessels were repeatedly cut with sterile ophthalmic scissors, and then placed in DMEM/F12 with 20% FBS at 37 °C and incubated a moist atmosphere of 5% CO2 at 37 ˚C. After 2 weeks, the well-grown cells show a typical "peak-valley" distribution. The cells were then passaged with 0.25% trypsin. When the cells were 70–80% confluent, the medium was replaced with DMEM and starved for 24 h. Then cells were treated with cobalt chloride (COCl2) at a final concentration of 100 μmol/L for 24 h to mimic hypoxia.
RNA extraction and quantitative RT-PCR
Total RNA from PASMCs was extracted using the FastPure Cell/Tissue Total RNA Isolation KIT V2(Vazyme, Nanjing, China) according to the manufacturer’s instructions. The cDNA synthesis was conducted using Hifair III 1st Strand cDNA Synthesis SuperMix (Yeasen Biotech, Shanghai, China). Using β-actin as a reference, we performed quantitative RT-PCR with Hieff qPCR SYBR Green Master Mix (Yeasen Biotech, Shanghai, China) in the LightCycler® 96 System (Roche Diagnostics, Mannheim, Germany). The 2−∆∆Ct method was applied to calculate the relative expression level of mRNA. The primer pairs used for amplification were as follows: PNISR F:5′-ATG TGG GAT CAA GGA GGA CAG-3′; R: 5′-CAG CCC AAT CAA TCT GGC TT-3′; HNRNPH1 F:5′-GGA GGC TAT GGA GGC TAT GAT-3′; R:5′-CCT GTT GTG CTC TGG AAA GTAG-3′; GAPDH F:5′-GGT GTG AAC CAT GAG AAG TAT GA-3′; R:5′GAG TCC TTC CAC GAT ACC AAAG–3′.
Statistical analysis
The GraphPad Prism 7 software (San Diego, CA, USA) were employed to analyze the data. Normal distribution measurement data were displayed as mean ± SD. The statistically significant differences between the PAH group and controls were performed by using Students t-test in GraphPad Prism 7. The construction of receiver operator characteristic (ROC) curve and the calculation of the area under the ROC curve (AUC) were finished in MedCalc Statistical Software version 14.8.1 (MedCalc Software Ltd, Ostend, Belgium). The statistical significance was set as P < 0.05.
Results
Identifications of DEGs
A total of 2298 differentially expressed genes were identified, with an adjusted P-value < 0.05 between PAH patients and controls. Compared with the controls, there were 1140 genes were upregulated and 1158 genes were downregulated in PAH. The volcano map of DEGs is shown in Fig. 2A. The heatmap for the top 50 DEGs is displayed in Fig. 2B.

Expression profile of DEGs. A The volcano map of DEGs. Black dots represent genes that are not differentially expressed between PAH patients and control patients. Blue indicates down-regulated genes, and red indicates up-regulated genes. B Heatmap of top 50 DEGs. The red color indicates the higher gene expression value while the green color indicates the lower gene expression
WGCNA analysis
The 2299 identified DEGs were further processed with the WGCNA package in R software, and a scale-free co-expression network (scale-free R2 > 0.8) was established using a soft thresholding power of 9. The soft thresholding power β was set at 24 in the subsequent analysis, because the scale independence reached 0.887 (Fig. 3A) and had a relatively good average connectivity. The DEGs were clustered into seven modules, purple, salmon, cyan, yellow, brown, midnight blue and grey, with a minimal module size > 30. The cluster dendrogram of the DEGs is shown in Fig. 3B. The correlation between each module and PAH was calculated and plotted (Fig. 3C). The results indicated that yellow (0.74, P < 0.0001) were the most positive modules related to PAH. Thus, the yellow module, including 597 DEGs, was considered as a key module correlated to PAH.

WGCNA of DEGs. A Estimation of the soft thresholding value for a scale-free coexpression network. B Cluster dendrogram of all DEGs. C Correlation between each module and patients or controls
Functional enrichment analysis
The 597 DEGs in the yellow module were used for GO and KEGG analysis with the Metascape tool. GO enrichment reults indicated that the yellow module genes were enriched in ‘transcription, DNA-templated’, ‘signal transduction’ and ‘positive regulation of transcription from RNA polymerase II promoter’ in BP terms (Fig. 4A). In terms of CC, the terms ‘cytoplasm’, ‘proteinaceous extracellular matrix’ and ‘lamellipodium’ were significantly enriched (Fig. 4B). The MF terms of the DEGs were ‘protein binding’, ‘metal ion binding’ and ‘zinc ion binding’ (Fig. 4C). Moreover, KEGG analysis demonstrated that the yellow module was enriched in ‘Pathways in cancer’, ‘PI3K-Akt signaling pathway’, and ‘Focal adhesion’ (Fig. 4D).

GSEA analysis
The distribution of the pathway gene sets on all gene expression data from the PAH patients and controls was explored using the GSEA software [19, 24]. The results showed that 27/50 gene sets were upregulated in the PAH patients, while 13 gene sets were significantly enriched with FDR < 25%. In the controls, 23/50 gene sets were upregulated, and 12 gene sets were highly enriched with FDR < 25%. The results of GSEA indicated that PAH patients enriched in ‘HALLMARK_MYC_TARGETS_V1’, ‘E2F_TARGETS’, ‘MTORC1_SIGNALING’, ‘DNA_REPAIR’, ‘G2M_CHECKPOINT’, and ‘GLYCOLYSIS’ pathway. The top six gene sets were shown in Fig. 5A–F.

Gene set enrichment analysis. A Enrichment plot of ‘MYC_TARGETS_V1’ with enrichment score 0.59, FDR q-value 0.0. B Enrichment plot of ‘E2F_TARGETS’ with enrichment score 0.47, FDR q-value 0.0. C Enrichment plot of ‘MTORC1_SIGNALING’ with enrichment score 0.65, FDR q-value 0.0. D Enrichment plot of ‘DNA_REPAIR’ with enrichment score 0.45, FDR q-value 0.0. E Enrichment plot of ‘G2M_CHECKPOINT’ with enrichment score 0.42, FDR q-value 0.0. F Enrichment plot of ‘GLYCOLYSIS’ with enrichment score 0.47, FDR q-value 0.0
PPI network construction, modular analysis, and hub gene analysis
The STRING database was used to construct the PPI network in order that we can explore the interaction of genes in the yellow module. Then, 0.7 was set as the threshold as the minimum required interaction score for constructing the STRING PPI network. As is shown in Fig. 6A, the PPI network comprised 230 nodes and 520 edges. Using the MCODE plug-in in Cytoscape, one module was determined. This Module (score = 7.0) included 7 nodes and 42 edges (Fig. 6B). The top ten hub genes obtained by MNC, MCC and Degree, in the cytohubba plug-in, are shown in Table 1. The overlapped hub genes among the three algorithms were verified by a Venn diagram (Fig. 6C), including VEGFA, KIT, PNISR and HNRNPH1.

PPI network and hub gene. A PPI network. B The most significant modules. C The overlapped hub genes from different algorithms
Validation of the hub genes
Base on the quantitative RT-qPCR experiment, the transcriptional changes of overlapped hub genes PNISR and HNRNPH1 were detected in the PASMCs from human by the method reported previously. The primary cultured PASMCs were divided into two groups: control group and CoCl2 treated group. The results indicated that the expression levels of PNISR and HNRNPH1 were both upregulated in the CoCl2 treated in comparison with those in controls (Fig. 7A, B), which was in line with the bioinformatics analysis, and the similar results were observed in rats (Additional file 1: Fig S1).To evaluate the capability of PNISR and HNRNPH1 to distinguish the CoCl2 treated from controls, ROC curves were performed. According to our data, the AUC of PNISR and HNRNPH1 were 0.815 (95% CI 0.690–0.905; P < 0.0001) and 0.744 (95% CI 0.611–0.850; P = 0.0005), respectively, showing that the identified hub genes PNISR and HNRNPH1 may be novel biomarkers of the PAH (Fig. 7C, D).

Validation of hub genes. A Relative mRNA level of PNISR in controls vs. PAH group. B Relative mRNA level of HNRNPH1 in controls versus PAH group. (**P < 0.01, ****P < 0.0001). C ROC curve for PNISR. D ROC curve for HNRNPH1
Discussion
PAH remains one of the serious and fatal lung disease with high morbidity and mortality worldwide [25]. PAH is often characterized by elevated pulmonary artery pressure and pulmonary vascular resistance, which finally leads to progressive right heart failure and death [26]. PAH patients might suffer severe exertional dyspnea, angina, fatigue, weakness, angina, presyncope, and syncope [27]. Without prompt intervention, most people may die from various symptoms. Pulmonary hypertension remains a global health problem [28]. So, it is particularly important to find new etiologies, effective diagnostic markers and treatment strategies.
In this study, a WGCNA on mRNA expression profile GSE117261 downloaded from the GEO database was performed. Based on the WGCNA analysis, all 2299 DEGs obtained by the “limma” package in R software were clustered into 7 modules. Then, 597 DEGs in the yellow module were found to be the most positive genes related to PAH (correlation score = 0.74, P < 0.0001), which were mainly enriched in ‘cytoplasm’ and closely related to ‘transcription, DNA-templated’, ‘protein binding’ and ‘pathways in cancer’. GSEA showed that the gene set ‘HALLMARK_MYC_TARGETS_V1’ was obviously enriched in the PAH group. Four hub genes, vascular endothelial growth factor A (VEGFA), v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), PNN Interacting Serine and Arginine Rich Protein (PNISR) and Heterogeneous Nuclear Ribonucleoprotein H1 (HNRNPH1) were determined based on the PPI network. Meanwhile, upregulatd expressions of PNISR and HNRNPH1 were validated by RT-qPCR.
Based on the GO analysis, the DEGs mainly consist of “Transcription, DNA-templated” in BP terms, which has been previously reported to be correlated to PAH [29]. The CC term “cytoplasm” and the MF term “protein binding” were both closely related to the transcription, which is an essential part of gene expression [30]. KEGG analysis revealed that the “Pathway in cancer” played a marked role in PAH, which was consistent with the previous demonstration demonstrated that the pathobiology of small vessels in severe PAH patients was quasi-neoplastic and cancer-like [31]. Meanwhile, the reliability of the DEGs obtained from the previous analysis was completely confirmed by results of the enrichment analysis.
By performing the GSEA analysis on the gene profile of GSE, many gene sets highly enriched in the PAH were found. The top six gene sets, including ‘HALLMARK_MYC_TARGETS_V1’, ‘E2F_TARGETS’, ‘MTORC1_SIGNALING’, ‘DNA_REPAIR’, ‘G2M_CHECKPOINT’, and ‘GLYCOLYSIS’ pathway, are cell cycle-related pathways, suggesting that genes involved in these pathways might contribute to cell proliferation. Among them, ‘HALLMARK_MYC_TARGETS_V1’ with an enrichment score of 0.59, was experimentally validated. This gene set involved 184 genes, including the well-known proliferating cell nuclear antigen (PCNA), minichromosome maintenance protein (MCM) and other cell proliferation markers [32]. Moreover, v-myc avian myelocytomatosis viral oncogene homolog (MYC) is a transcription factor known to regulate various human genes, promoting cell growth and proliferation [33], regulating apoptosis by altering pro- and anti-apoptotic members of the BCL-2 family and activating telomerase, controling the angiogenesis by regulating the expression levels of VEGF [34].
More and more studies showed that cell proliferation was one of the pathophysiological mechanisms of pulmonary arterial hypertension [35]. Inhibition of the proliferation of pulmonary artery smooth muscle cells can effectively ameliorate pulmonary arterial hypertension [36]. We are delighted to find that cell proliferation is enriched in PAH in accordance with the previous findings.
Based on previous studies, our work provides a new insight into the underlying pathogenesis of PAH. Dong H et al. identified CSF3R, NT5E, ANGPT2, FGF7 and CXCL9 as candidate biomarkers of PAH, and ruxolitinib might exert promising therapeutic action for PAH [37]. Qiu X et al. performed a WGCNA on GSE15197 and found that 11 real hub genes, including EP300, MMP2, CDH2, CDK2, GNG10, ALB, SMC2, DHX15, CUL3, BTBD1, and LTN1, were over-expressed in IPAH [38]. Contrastly, Liu J et al. suggested that 10 hub genes were determined via Cytohubba and a crucially ceRNA network was identified, including 14 LncRNAs, 2 miRNAs, and 3 mRNAs [39]. Interestingly, Li Q et al. confirmed that 9 hub genes related to PAH, particularly the PLK4 and SMC2 genes, providing a deeper understanding of physiopathologic of PAH [29]. Farha S et al. inferred that inhibition of KIT progenitor could improve remodeling and proliferation in PAH. Imatinib, a tyrosine kinase inhibitor that targets c-KIT, has been shown to be beneficial to PAH patients due to its inhibition of proliferation in hematopoietic progenitors and mast cells [40]. Liu J et al. found that VEGF initiated vascular remodeling resulting in PAH [41]. However, to our knowledge, research on the role of PNISR and HNRNPH1 in PAH remains limited.
PNISR is a newly identified serine-arginine (SR) protein in human that co-purifies with pinin, which is rarely reported [42]. Sinclair PB et al. suggested that abnormal expressions of GRIK2 and PNISR were associated with proliferation in some lymphoid leukemias [43]. HNRNPH1, a core member of the heterogeneous nuclear ribonucleo-proteins family, frequently upregulated in various cancer cells and contributed to tumorigenesis [44]. Elevated expression of HNRNPH1 was found in acute myeloid leukemia (AML), and knockdown of HNRNPH1 alleviated cell proliferation [45]. And the upregulated expressions of PNISR and HNRNPH1 in PAH were found in our study. According to the ROC curve, the values of the AUC of PNISR and HNRNPH1 were 0.815 and 0.744, respectively. The AUC result indicated that PNISR and HNRNPH1 had a powerful ability to discriminate PAH from the controls. Together, PNISR and HNRNPH1 were considered as candidate biomarkers of PAH. Nevertheless, further research are required to investigate the role of PNISR and HNRNPH1 on the development of PAH, and the expansion of the sample size is needed to validate the efficacy of PNISR and HNRNPH1 as biomarkers for PAH.
Conclusions
Conclusively, PNISR and HNRNPH1 were determined to be potential biomarkers in PAH. Our results indicate that PNISR and HNRNPH1 participate in the development of PAH and serve as potential diagnosis and therapeutic targets for PAH.
Availability of data and materials
The dataset analyzed in this study can be derived from public repositories: GSE117261 dataset (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=gse117261).
References
Klinger JR. novel pharmacological targets for pulmonary arterial hypertension. Compr Physiol. 2021;11(4):2297–349.
Barnett CF, Alvarez P, Park MH. Pulmonary arterial hypertension: diagnosis and treatment. Cardiol Clin. 2016;34(3):375–89.
Zhang JR, Ouyang X, Hou C, Yang QF, Wu Y, Lu WJ, Liu CL, Yang K, Zhong NS, Wang J, et al. Natural ingredients from Chinese materia medica for pulmonary hypertension. Chin J Nat Med. 2021;19(11):801–14.
Bisserier M, Pradhan N, Hadri L. Current and emerging therapeutic approaches to pulmonary hypertension. Rev Cardiovasc Med. 2020;21(2):163–79.
Zhao E, Xie H, Zhang Y. Identification of differentially expressed genes associated with idiopathic pulmonary arterial hypertension by integrated bioinformatics approaches. J Comput Biol. 2021;28(1):79–88.
Wang T, Zheng X, Li R, Liu X, Wu J, Zhong X, Zhang W, Liu Y, He X, Liu W, et al. Integrated bioinformatic analysis reveals YWHAB as a novel diagnostic biomarker for idiopathic pulmonary arterial hypertension. J Cell Physiol. 2019;234(5):6449–62.
Zheng JN, Li Y, Yan YM, Shi H, Zou TT, Shao WQ, Wang Q. Identification and validation of key genes associated with systemic sclerosis-related pulmonary hypertension. Front Genet. 2020;11:816.
Bai KH, He SY, Shu LL, Wang WD, Lin SY, Zhang QY, Li L, Cheng L, Dai YJ. Identification of cancer stem cell characteristics in liver hepatocellular carcinoma by WGCNA analysis of transcriptome stemness index. Cancer Med. 2020;9(12):4290–8.
Stearman RS, Bui QM, Speyer G, Handen A, Cornelius AR, Graham BB, Kim S, Mickler EA, Tuder RM, Chan SY, et al. Systems analysis of the human pulmonary arterial hypertension lung transcriptome. Am J Respir Cell Mol Biol. 2019;60(6):637–49.
Romanoski CE, Qi X, Sangam S, Vanderpool RR, Stearman RS, Conklin A, Gonzalez-Garay M, Rischard F, Ayon RJ, Wang J, et al. Transcriptomic profiles in pulmonary arterial hypertension associate with disease severity and identify novel candidate genes. Pulm Circ. 2020;10(4):2045894020968531.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7): e47.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9:559.
The Gene Ontology C. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47(1):D330–8.
Kanehisa M, Sato Y, Furumichi M, Morishima K, Tanabe M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 2019;47(D1):D590–5.
Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–7.
Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30.
Kanehisa M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019;28(11):1947–51.
Kanehisa M, Furumichi M, Sato Y, Ishiguro-Watanabe M, Tanabe M. KEGG: integrating viruses and cellular organisms. Nucleic Acids Res. 2021;49(D1):D545–51.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–50.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808-815.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003;4:2.
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(Suppl 4):S11.
Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–73.
Hensley MK, Levine A, Gladwin MT, Lai YC. Emerging therapeutics in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2018;314(5):L769–81.
Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, Nicolls MR, Olschewski AJ, Pullamsetti SS, Schermuly RT, Stenmark KR et al: Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019, 53(1).
Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, VonkNoordegraaf A, Beghetti M, et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: the joint task force for the diagnosis and treatment of pulmonary hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37(1):67–119.
Galie N, Channick RN, Frantz RP, Grunig E, Jing ZC, Moiseeva O, Preston IR, Pulido T, Safdar Z, Tamura Y et al: Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J 2019, 53(1).
Li Q, Meng L, Liu D. Screening and identification of therapeutic targets for pulmonary arterial hypertension through microarray technology. Front Genet. 2020;11:782.
Gegenfurtner FA, Zisis T, Al Danaf N, Schrimpf W, Kliesmete Z, Ziegenhain C, Enard W, Kazmaier U, Lamb DC, Vollmar AM, et al. Transcriptional effects of actin-binding compounds: the cytoplasm sets the tone. Cell Mol Life Sci. 2018;75(24):4539–55.
Rai PR, Cool CD, King JA, Stevens T, Burns N, Winn RA, Kasper M, Voelkel NF. The cancer paradigm of severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178(6):558–64.
Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The molecular signatures database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–25.
Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F. The c-Myc target gene network. Semin Cancer Biol. 2006;16(4):253–64.
Xu J, Chen Y, Olopade OI. MYC and breast cancer. Genes Cancer. 2010;1(6):629–40.
Schermuly RT, Ghofrani HA, Wilkins MR, Grimminger F. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol. 2011;8(8):443–55.
Zhang L, Wang Y, Wu G, Rao L, Wei Y, Yue H, Yuan T, Yang P, Xiong F, Zhang S, et al. Blockade of JAK2 protects mice against hypoxia-induced pulmonary arterial hypertension by repressing pulmonary arterial smooth muscle cell proliferation. Cell Prolif. 2020;53(2): e12742.
Dong H, Li X, Cai M, Zhang C, Mao W, Wang Y, Xu Q, Chen M, Wang L, Huang X. Integrated bioinformatic analysis reveals the underlying molecular mechanism of and potential drugs for pulmonary arterial hypertension. Aging (Albany NY). 2021;13(10):14234–57.
Qiu X, Lin J, Liang B, Chen Y, Liu G, Zheng J. Identification of hub genes and MicroRNAs associated with idiopathic pulmonary arterial hypertension by integrated bioinformatics analyses. Front Genet. 2021;12: 667406.
Liu J, Sun Y, Zhu B, Lin Y, Lin K, Sun Y, Yao Z, Yuan L. Identification of a potentially novel LncRNA-miRNA-mRNA competing endogenous RNA network in pulmonary arterial hypertension via integrated bioinformatic analysis. Life Sci. 2021;277: 119455.
Farha S, Dweik R, Rahaghi F, Benza R, Hassoun P, Frantz R, Torres F, Quinn DA, Comhair S, Erzurum S, et al. Imatinib in pulmonary arterial hypertension: c-Kit inhibition. Pulm Circ. 2014;4(3):452–5.
Liu J, Wang W, Wang L, Chen S, Tian B, Huang K, Corrigan CJ, Ying S, Wang W, Wang C. IL-33 initiates vascular remodelling in hypoxic pulmonary hypertension by up-regulating HIF-1alpha and VEGF expression in vascular endothelial cells. EBioMedicine. 2018;33:196–210.
Zimowska G, Shi J, Munguba G, Jackson MR, Alpatov R, Simmons MN, Shi Y, Sugrue SP. Pinin/DRS/memA interacts with SRp75, SRm300 and SRrp130 in corneal epithelial cells. Invest Ophthalmol Vis Sci. 2003;44(11):4715–23.
Sinclair PB, Sorour A, Martineau M, Harrison CJ, Mitchell WA, O’Neill E, Foroni L. A fluorescence in situ hybridization map of 6q deletions in acute lymphocytic leukemia: identification and analysis of a candidate tumor suppressor gene. Cancer Res. 2004;64(12):4089–98.
Garneau D, Revil T, Fisette JF, Chabot B. Heterogeneous nuclear ribonucleoprotein F/H proteins modulate the alternative splicing of the apoptotic mediator Bcl-x. J Biol Chem. 2005;280(24):22641–50.
Lefave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, Pan YX, Cartegni L. Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J. 2011;30(19):4084–97.
Acknowledgements
The authors would like to thank the editor and reviewers for their valuable comments and acknowledge GEO database for providing their platform and contributors for uploading their meaningful datasets.
Funding
This work was supported by the grants from the National Natural Science Foundation of China (82170355 and 81873537) and Joint Funds for the Innovation of Science and Technology, Fujian province (2019Y9124).
Author information
Authors and Affiliations
Contributions
WW and LX designed the research. WW performed the research and data collection. AC, SL, QW, GL, and LL contributed new reagents or analytic tools. WW, LX, AC and GL analyzed data. WW wrote the paper. All authors read and approved the fnal manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
GEO belongs to public databases. The patients involved in the database have obtained ethical approve. Users can download data for free for research and publish relevant articles. Our study was approved by the Branch for Medical Research and Clinical Technology Application, Ethics Committee of the First Affiliated Hospital of Fujian Medical University (Approval No. MRCTA, ECFAH of FMU[2021]483). Informed consent was obtained from all patients involved. Study was carried out in accordance with ethical guidelines of Fujian Medical University. Informed consent was obtained from participants/legal guardians.
Consent for publication
Not applicable.
Competing interests
The authors declare that there are no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Validation of hub genes in rat PASMCs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, W., Chen, A., Lin, S. et al. The identification and verification of hub genes associated with pulmonary arterial hypertension using weighted gene co-expression network analysis. BMC Pulm Med 22, 474 (2022). https://doi.org/10.1186/s12890-022-02275-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-022-02275-6