
Abstract
Background
Chronic obstructive pulmonary disease (COPD) is a disease that causes obstructed airways and abnormal inflammatory responses in the lungs. Early growth response 3 (EGR3) has been revealed to play a vital role in the regulation of the inflammatory response in certain diseases. We aimed to explore the role of EGR3 and its upstream mechanism in COPD.
Methods and result
In the present study, 16HBE cells were treated with cigarette smoke extract (CSE) to mimic the inflammatory response in vitro. RT-qPCR revealed that the expression of EGR3 was upregulated in lungs from COPD patients. EGR3 expression in 16HBE cells was increased by CSE treatment. Moreover, flow cytometry analysis and western blot analysis showed that EGR3 downregulation inhibited 16HBE cell apoptosis. EGR3 silencing decreased the protein levels of IL-6, TNF-α, IL-1β and COX2 in CSE-stimulated 16HBE cells. In addition, EGR3 was targeted by microRNA-200c-3p (miR-200c-3p) in 16HBE cells. MiR-200c-3p expression was significantly decreased in lung tissues from COPD patients compared to that in healthy controls. Furthermore, miR-200c-3p bound to lncRNA X-inactive specific transcript (XIST) in 16HBE cells. Additionally, XIST expression was elevated in lung tissues from COPD patients. Rescue assays indicated that EGR3 overexpression counteracted the effects of XIST downregulation on apoptosis and inflammation in CSE-stimulated 16HBE cells.
Conclusion
The XIST/miR-200c-3p/EGR3 axis facilitated apoptosis and inflammation in CSE-stimulated 16HBE cells. These findings may provide novel insight for treating COPD by alleviating lung inflammation.
Similar content being viewed by others

Introduction
Chronic obstructive pulmonary disease (COPD) is a disease that causes obstructed airways and abnormal inflammatory responses in the lungs [1, 2]. COPD affects approximate 328 million people worldwide each year and causes 3.5–4 million deaths [3]. Smoking-induced chronic inflammation, tissue destruction and inhibition of tissue repair in the lungs are the major factors for COPD [4]. The inhalation of cigarette smoke activates pattern-recognition receptors, stimulating an innate immune response that activates the airway epithelial cells [5]. Activation of these cells triggers the release of various proinflammatory cytokines that recruit neutrophils perpetuate chronic inflammation in COPD [6, 7].
Early growth response 3 (EGR3), also known as PILOT, is a member of the EGR family of C2H2-type zinc-finger proteins [8]. Many studies have revealed that EGR3 plays a critical role in the inflammatory response in various pathogeneses [9, 10]. The transcription factor EGR3 is essential for the proliferation and the inflammatory response of B and T cells [10]. EGR3 activates the transcription of proinflammatory cytokines, interleukin (IL)-6 and IL-8, to facilitate the inflammatory response in prostate cancer [11]. EGR3 acts as an oncogene in lung cancer [12], and COPD has been indicated to worsen lung cancer prognosis [13]. Considering the role of EGR3 in inflammation-associated diseases and in lung cancer as mentioned above, we hypothesized that EGR3 exerts a significant effect in COPD.
Recently, the competing endogenous RNA (ceRNA) hypothesis has been widely proposed to play a pivotal role in COPD [14, 15]. The ceRNA hypothesis indicates that long noncoding RNA (lncRNA) transcripts containing miRNA-binding sites can stabilize mRNA expression by competing for shared microRNAs (miRNAs) [16]. MiRNAs are small noncoding RNAs that modulate gene expression by binding to the 3′ untranslated region (3′UTR) of target messenger RNAs (mRNAs) to degrade mRNAs [17, 18]. We hypothesized that EGR3 is regulated by the lncRNA mediated ceRNA pattern in COPD.
In this study, the expression profile of EGR3 in lung tissues of COPD patients and its function in inflammatory response of an in vitro COPD model were investigated. Moreover, the upstream ceRNA mechanism underlying EGR3 mediated by lncRNA was explored. Our research may provide a potential novel direction for the clinical treatment of COPD.
Materials and methods
Patients and specimens
Lung specimens were collected from fifty-five patients with solitary non-small cell lung cancer receiving lobectomy or pneumonectomy combined with resection of a part of left atrium at Tianjin First Central Hospital between July 2016 and July 2018. The samples were divided into the following three groups: nonsmokers without COPD (n = 7; 5 men and 2 women; age range, 36–73 years; mean age, 55.1 ± 5.9 years old), smokers without COPD (n = 22; 16 men and 6 women; age range, 34–70 years; mean age, 52.6 ± 5.5 years old), and smokers with COPD (n = 26; 21 men and 5 women; age range, 31–74 years; mean age, 50.7 ± 5.3 years old). COPD was diagnosed through systemic physiological examinations according to the standard of the Global Initiative for Chronic Obstruct Lung Disease. Patients in the “smokers with COPD” group were first diagnosed with COPD and no other severe clinical disorders were observed. All nonsmokers without COPD received systemic physiological examinations and all parameters were within normal ranges. The smokers without COPD had a normal lung function at the time of admission and a history of smoking for 10.1–17.5 years. Patients were excluded if they suffered from asthma or other obstructive lung diseases. Written informed consents were obtained from all participants. The Ethics Committee of Tianjin First Central Hospital approved our study (approval number: 2020-011).
Immunohistochemistry (IHC)
The airway tissues were inactivated with 3% hydrogen peroxide for 10 min at room temperature and blocked with 5% bovine serum albumin for 20 min followed by incubation with anti-EGR3 polyclonal antibody (1:200, ab232820, Abcam) at 4 °C overnight. Next, the sections were incubated with the horseradish peroxide-conjugated secondary antibody at room temperature for 2 h. Subsequently, EGR3 protein was visualized using diaminobenzidine and counterstained with hematoxylin. Images of stained airway tissues were photographed with an inverted fluorescence microscope (Olympus, Japan) and analyzed with Image Pro Plus 6.0 software.
Cell culture and treatment
Exposure to cigarette smoke extract (CSE) can cause an abnormal inflammatory response in the small airways and alveoli, thereby accelerating the apoptosis of bronchial epithelial cells [19]. CSE-treated bronchial epithelial cells are widely used as an in vitro model of COPD [20,21,22]. The normal human bronchial epithelial cell line 16HBE was purchased from American Type Culture Collection (ATCC; USA) and cultured in RPMI-1640 medium (Invitrogen, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen) at 37 °C with 5% CO2. For cell treatment, different concentrations (1%, 2%, 3% and 4%) of cigarette smoke extract (CSE) were prepared using 10 cigarettes (Suyan, Jiangsu Cigarettes Company, Jiangsu, China) as previously described [23]. In brief, the smoke from 10 cigarettes (Furong, Changde Cigarette Company, Hunan, China) was bubbled using 25-mL media. The suspension was titrated to pH 7.4, filter-sterilized, and regarded as 100% CES. The CSE sample was diluted with phosphate-buffered saline (PBS) to concentrations of 1%, 2%, 3% and 4%, and then stored at − 80 °C. The 16HBE cells were treated with CSE for 24 h. Cells in the control group were treated with the same dose of PBS.
Cell transfection
Short hairpin RNA against XIST (sh-XIST) or EGR3 (sh-EGR3) was utilized to knock down XIST or EGR3, respectively. sh-NC served as the negative control for sh-XIST or sh-EGR3. Coding region of EGR3 was inserted into the pcDNA3.1 vector to overexpress EGR3 and empty pcDNA3.1 vector (Vector) was used as the negative control. MiR-200c-3p mimics (miR-200c-3p) was used for the overexpression of miR-200c-3p and NC mimics was regarded as the negative control. All vectors were constructed by GenePharma (Shanghai, China). 16HBE cells (1 × 105 cells/well) were seeded in 24-well plates with 500 μL of RPMI-1640 medium (Invitrogen) in each well. When the cells reached 40–60% confluence, the aforementioned vectors were transfected into cells at a final concentration of 50 nM using Lipofectamine® 2000 reagent (Invitrogen) at 37 °C and 5% CO2 for 48 h under the manufacturer’s instructions. Additionally, the related sequences of oligonucleotide used in this research were shown in Additional file 1: Table S1.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
The airway tissues were dissected, and relative RNA expression was detected in airway tissues. Total RNA was extracted from airway tissues and 16HBE cells with TRIzol reagent (Takara, Japan). Reverse transcription for EGR3 and XIST was performed with the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, USA). Real-time PCR was performed using SYBR Green Real-Time PCR MasterMix (Toyobo, Japan) with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal control. For the quantification of miR-200c-3p, total miRNA was extracted from tissues and cells using the miRNeasy RNA Isolation Kit (Qiagen, USA). Total extract was reverse transcribed into cDNA using the miScript RT Kit (Qiagen) with RNU6 (U6) as the internal control. Real-time PCR was performed with the Bio-Rad CFX96 qPCR system (Hercules, USA), and expression was calculated with the 2−△△CT method. The related sequences of primers were shown in Additional file 1: Table S1.
Enzyme-linked immunosorbent assay (ELISA)
Concentrations of IL-6, TNF-α, COX2 and IL-1βin culture supernatant collected from 16HBE cells under different treatments were determined using commercial ELISA kits (R&D Systems, USA).
Bioinformatics analysis
MiRNAs that potentially target EGR3 and lncRNAs that potentially bind to miR-200c-3p were predicted from starBase 2.0 website (http://starbase.sysu.edu.cn/). MiR-200c-3p and miR-429 were predicted to target EGR3 according to the overlapping prediction results of RNA22, PicTar and TargetScan tools. LncRNAs (XIST, RRN3P2, AC120036.4) which potentially bind with miR-200c-3p were predicted under the conditions of medium stringency of CLIP data and low stringency of degradome data. Putative binding site between XIST and miR-200c-3p was obtained from starBase 2.0 website. Putative binding site between EGR3 and miR-200c-3p was predicted by TargetScan 7.2 website (http://www.targetscan.org/vert_72/).
RNA immunoprecipitation (RIP) assay
RNA immunoprecipitation (RIP) assays were performed with a Magna RNA Immunoprecipitation Kit (Millipore, MA, USA). 16HBE cells were lysed in RIPA buffer containing magnetic beads conjugated with anti-Ago2 or anti-IgG. Argonaute 2 (Ago2) is a catalytic component of the RNA-induced silencing complexes (RISCs) [24]. MiRNA precursors were transcribed, processed into mature miRNAs and loaded onto Ago2 protein to form the RISC. Thus, miRNAs and their bound RNAs can be identified by immunoprecipitation of Ago2. Then, the immunoprecipitated RNAs were isolated by TRIzol reagent, and the enrichment of EGR3, miR-200c-3p and XIST was analyzed by RT-qPCR.
Luciferase reporter assay
The XIST sequence or the 3’UTR fragment of EGR3 containing the predicted binding site for miR-200c-3p was subcloned into a pmirGLO-luciferase Target Expression Vector to construct the XIST wild-type (pmirGLO-XIST-Wt) vector or the EGR3 3’UTR wild-type (pmirGLO-EGR3 3’UTR-Wt) vector, respectively. XIST sequence or EGR3 3’UTR was directed-mutated by Genepharma and were inserted into the pmirGLO vector to construct the pmirGLO-EGR3 3’UTR-Mut or pmirGLO-XIST-Mut plasmids. 16HBE cells were cotransfected with pmirGLO-EGR3 3’UTR-Wt vector (or pmirGLO-XIST-Wt vector), pmirGLO-EGR3 3’UTR-Mut vector (or pmirGLO-XIST-Mut vector) and miR-200c-3p mimics (or NC mimics) using Lipofectamine 2000 for 48 h. A dual luciferase assay kit (Promega) was applied to investigate luciferase activity.
Flow cytometry analysis
Flow cytometry analysis was applied to detect apoptosis of 16HBE cells. After being transfected with the indicated plasmids for 48 h, the cells were rinsed with PBS twice followed by resuspension in binding buffer. Next, the cells were stained with Annexin V-Fluorescein Isothiocyanate (FITC) and propidium iodide (PI) (BD Biosciences, USA) in a dark room for 20 min. Apoptosis was measured by flow cytometry using a BD FACSCalibur system (BD Biosciences). Q1 represents living cells; Q2 represents early apoptotic cells; Q3 represents late apoptotic cells; Q4 represents necrotic cells. Cell apoptosis rate = number of cells in Q2 + Q3/number of cells in all quadrants.
Western blot analysis
Total proteins were extracted from 16HBE cells with radioimmunoprecipitation assay (RIPA) buffer (Beyotime, Jiangsu, China) containing protease inhibitors (Beyotime). Protein concentrations were determined with the bicinchoninic acid (BCA) Protein Assay Kit (Beyotime). Equal amounts of proteins were separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and then transferred onto nitrocellulose membranes (Millipore, USA). After blocking with 5% nonfat milk, the membranes were incubated with specific primary antibodies, including anti-EGR3 (ab221711), anti-Bax (ab32503), anti-Bcl-2 (ab196495), anti-cleaved caspase-3 (ab32042), anti-IL-6 (ab233706), anti-TNF-α (ab183218), anti-COX-2 (ab179800), anti-IL-1β (ab2105) and anti-GAPDH (ab181602). After washing with PBS, the membranes were incubated with goat anti-mouse IgG (ab150077) and analyzed using an Odyssey infrared scanner (Li-Cor, USA). All antibodies were purchased from Abcam (Cambridge, USA).
Statistical analysis
Data were processed using SPSS 17.0 statistical software and are shown as the mean ± standard deviation. Kolmogorov–Smirnov tests were performed for checking data normality. All data were normally distributed. Student’s t-test was applied for statistical comparison between two groups, while differences among more than two groups were estimated using one-way analysis of variance followed by Tukey’s post hoc test. Pearson’s correlation analysis was used to analyze the correlations between the expression of EGR3 and miR-200c-3p, miR-200c-3p and XIST, and EGR3 and XIST in the lung tissues of smokers. p < 0.05 was considered statistically significant.
Results
EGR3 was upregulated in COPD lungs
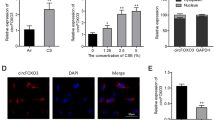
First, we investigated the expression of EGR3 in the clinical lung samples. The data revealed that EGR3 was upregulated in the lung tissues of smokers compared with that in the lung tissues of nonsmokers. EGR3 expression was higher in the lung tissues of smokers with COPD than smokers without COPD (Fig. 1a). Results of IHC staining assay of airway tissues revealed that EGR3 was mainly located in the cytoplasm of bronchial epithelial cells. EGR3 expression was significantly increased in the smoker group compared with nonsmoker group, and increased in COPD group compared with non-COPD group (Fig. 1b). Moreover, a normal human bronchial epithelial cell line (16HBE) was exposed to varying concentrations (1%, 2%, 3% and 4%) of CSE for 24 h. We found that EGR3 expression was increased in CES-treated 16HBE cells in a dose-dependent manner (Fig. 1c). Additionally, EGR3 expression was elevated in 16HBE cells exposed to 2% CSE in a time-dependent manner (Fig. 1d). All these experimental results suggested that the in vitro cellular model was successfully established, and 2% CSE treatment for 24 h was used for the following assays.

EGR3 was upregulated in COPD lungs. a RT-qPCR was used to detect the expression of EGR3 in lungs from nonsmokers (n = 7), smokers without COPD (n = 22) and smokers with COPD (n = 26). b IHC staining of EGR3 in airway tissues of nonsmokers, smokers without COPD, smokers with COPD. c The expression of EGR3 in 16HBE cells treated with different concentrations of CSE (1, 2, 3, and 4%) for 24 h was evaluated by RT-qPCR (n = 3). d The expression of EGR3 in 16HBE cells exposed to 2.5% CSE at 0, 12, 24, 36 and 48 h was measured by RT-qPCR (n = 3). *p < 0.05, **p < 0.01
EGR3 downregulation inhibited apoptosis and the inflammatory response in CSE-treated cells
We then conducted loss-of-function assays to explore the function of EGR3 in CSE-treated cells. EGR3 expression was downregulated by transfection with sh-EGR3 in basal 16HBE cells and CSE-stimulated 16HBE cells (Fig. 2a, b). Moreover, flow cytometry analysis showed that CSE treatment increased the apoptosis rate of 16HBE cells, and EGR3 downregulation decreased the apoptosis rate of CSE-treated 16HBE cells (Fig. 2c). Additionally, the levels of apoptosis-related proteins were measured. The results revealed that Bax and cleaved caspase-3 protein levels were increased, and the Bcl-2 protein level was decreased by CSE treatment in 16HBE cells, and EGR3 downregulation counteracted these effects (Fig. 2d). Moreover, the concentrations and protein levels of proinflammatory factors (IL-6, TNF-α, IL-1β and COX2) in 16HBE cells were increased by CSE treatment and repressed by knockdown of EGR3 in the supernatants of CSE-treated cells (Fig. 2e, f).

EGR3 downregulation inhibited apoptosis and the inflammatory response in CSE-treated cells. a The transfection efficacy of sh-EGR3 and sh-NC in 16HBE cells was tested by RT-qPCR (n = 3). b The expression of EGR3 in CSE-treated 16HBE cells under indicated treatment was analyzed by RT-qPCR (n = 3). c Flow cytometry analysis was applied to assess apoptosis in CSE-treated cells (n = 3). d Western blot analysis was used to measure the levels of apoptosis-related proteins (Bax, Bcl-2 and cleaved caspase-3) in CSE-treated 16HBE cells (n = 3). e ELISA was used to measure the concentrations of proinflammatory factors (IL-6, TNF-α, IL-1β and COX2) in CSE-treated 16HBE cells (n = 3). f The protein levels of IL-6, TNF-α, IL-1β and COX2 in CSE-treated 16HBE cells were measured by western blot analysis (n = 3). **p < 0.01, ***p < 0.001
EGR3 was a downstream target of miR-200c-3p
As the role of EGR3 in COPD was corroborated above, the upstream mechanism underlying EGR3 was explored. According to the starBase database (http://starbase.sysu.edu.cn/), miR-429 and miR-200c-3p were predicted to possess potential binding sites on the EGR3 3’UTR (Fig. 3a). Thereafter, RT-qPCR showed that miR-200c-3p expression was lower in the lung tissue of smokers compared to that of nonsmokers. Moreover, miR-200c-3p expression was lower in smokers with COPD than in smokers without COPD (Fig. 3b). In addition, a RIP assay was used to assess whether miR-200c-3p can bind with EGR3 in 16HBE cells. From the results, we observed that EGR3 and miR-200c-3p were both enriched in the Ago2 group but not in the IgG group (Fig. 3c), implying that miR-200c-3p targeted EGR3 in 16HBE cells. Next, miR-200c-3p was overexpressed by transfection with miR-200c-3p mimics into 16HBE cells (Fig. 3d). The results of the luciferase reporter assay revealed that the overexpression of miR-200c-3p decreased the luciferase activity of EGR3-Wt but not that of EGR3-Mut in 16HBE cells (Fig. 3e, f). Moreover, EGR3 expression was negatively correlated with miR-200c-3p expression in the lung tissues of 48 smokers (Fig. 3g). Furthermore, RT-qPCR and western blot analysis indicated that miR-200c-3p negatively regulated EGR3 mRNA and protein levels in 16HBE cells (Fig. 3h).

EGR3 was a downstream target of miR-200c-3p. a Venn diagram showing the potential miRNAs possessing binding sites on the EGR3 3’UTR. b RT-qPCR was used to examine the expression of miR-200c-3p and miR-429 in lungs from nonsmokers (n = 7), smokers without COPD (n = 22), and smokers with COPD (n = 26). c RIP assay was conducted to reveal the relative enrichment of miR-200c-3p and EGR3 precipitated by Ago2 normalized to that by IgG in 16HBE cells (n = 3). d RT-qPCR was used to measure miR-200c-3p expression in 16HBE cells transfected with NC mimics or miR-200c-3p mimics (n = 3). e The starBase database predicted the potential binding site of miR-200c-3p on the EGR3 3’UTR. f Luciferase reporter assay was used to assess the binding ability of miR-200c-3p and EGR3 in 16HBE cells (n = 3). g The correlation between EGR3 and miR-200c-3p expression in lung tissues of smokers (n = 48) was analyzed by Pearson’s correlation analysis. h The mRNA and protein levels of miR-200c-3p in response to miR-200c-3p overexpression in 16HBE cells (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001
XIST bound with miR-200c-3p to regulate EGR3
We wondered whether EGR3 and miR-200c-3p are involved in the ceRNA network in 16HBE cells. The starBase database was used, and three lncRNAs (XIST, RRN3P2 and AC120036.4) were found to bind with miR-200c-3p. As shown in Fig. 4a, only XIST expression was significantly higher in the lung tissues of smokers than non-smokers, whereas the other two lncRNAs presented no differential expression difference in the lung tissues of smokers and nonsmokers. Furthermore, XIST expression was significantly higher in the lung tissues of smokers with COPD than smokers without COPD. The interaction between miR-200c-3p and XIST was then verified by RIP assay. Abundant enrichment of miR-200c-3p and XIST in 16HBE cells was observed in the Ago2 group but not in the IgG group (Fig. 4b). The binding sequence between XIST and miR-200c-3p was revealed in Fig. 4c. A luciferase reporter assay suggested that the luciferase activity of XIST-Wt was decreased in the miR-200c-3p mimic group compared to the NC mimic group, and the luciferase activity of XIST-Mut showed no evident change in either group (Fig. 4d). Pearson’s correlation analysis showed that miR-200c-3p expression was negatively associated with XIST expression and that EGR3 expression was positively associated with XIST expression in the lung tissues of 48 smokers (Fig. 4e, f). Additionally, in response to XIST silencing in 16HBE cells, the levels of both XIST and EGR3 were decreased, and the level of miR-200c-3p is not significantly affected (Fig. 4g). Furthermore, the protein level of EGR3 was reduced by knockdown of XIST (Fig. 4h). After silencing of XIST, the enrichment of EGR3 in the RISCs was increased, indicating the increase of EGR3 binding to miR-200c-3p (Fig. 4I).

MiR-200c-3p interacted with XIST. a The levels of XIST, RRN3P2 and AC120036.4 in lung tissues from nonsmokers (n = 7), smokers without COPD (n = 22) and smokers with COPD (n = 26). b RIP assay was used to assess the relative enrichment of XIST and miR-200c-3p precipitated by Ago2 normalized to that by IgG in 16HBE cells (n = 3). c The starBase database showed the potential binding site for miR-200c-3p and XIST. d The binding ability between miR-200c-3p and XIST in 16HBE cells was verified by luciferase reporter assay (n = 3). e and f, Pearson’s correlation analysis demonstrated the correlation between XIST and EGR3 (or miR-200c-3p) expression in lung tissues of smokers (n = 48). g The transfection efficacy of sh-XIST and sh-NC as well as the levels of miR-200c-3p and EGR3 in 16HBE cells under XIST knockdown (n = 3). h The effect of XIST on EGR3 protein levels in 16HBE cells (n = 3). i RIP assay was conducted to reveal the relative enrichment of EGR3 precipitated by Ago2 normalized to that by IgG in 16HBE cells with silencing of XIST. *p < 0.05, **p < 0.01, ***p < 0.001
EGR3 overexpression reversed the XIST downregulation-mediated suppression of apoptosis and the inflammatory response in CSE-treated 16HBE cells
Subsequently, we explored the role of XIST in CSE-treated 16HBE cells and investigated whether EGR3 participates in XIST-mediated cellular behaviors. First, we transfected CSE-treated 16HBE cells with pcDNA3.1 or pcDNA3.1/EGR3. The results suggested that EGR3 mRNA and protein levels were successfully overexpressed by transfection with pcDNA3.1/EGR3 in 16HBE cells (Fig. 5a). Additionally, flow cytometry analysis showed that EGR3 overexpression reversed the decrease of the cell apoptosis rate induced by XIST silencing in CSE-stimulated 16HBE cells (Fig. 5b). Moreover, the increase in Bcl-2 protein levels and the decrease in Bax and cleaved caspase-3 protein levels induced by XIST downregulation were rescued by EGR3 overexpression in CSE-stimulated 16HBE cells (Fig. 5c). In addition, the decreased concentrations and protein levels of IL-6, TNF-α, IL-1β and COX2 induced by XIST knockdown were reversed by EGR3 overexpression in CSE-treated 16HBE cells (Fig. 5d, e).

EGR3 reversed the XIST downregulation-mediated suppression of apoptosis and inflammation in CSE-stimulated cells. a RT-qPCR and western blot analysis were used to evaluate the transfection efficacy of pcDNA3.1 or pcDNA3.1/EGR3 in 16HBE cells (n = 3). b Flow cytometry analysis showed the apoptosis of CSE-treated 16HBE cells in each group (n = 3). c Western blot analysis was used to measure the protein levels of Bax, Bcl-2 and cleaved caspase-3 in CSE-treated 16HBE cells under the indicated transfection conditions (n = 3). d The concentrations of IL-6, TNF-α, IL-1β and COX2 in CSE-treated 16HBE cells under the indicated treatments were measured by ELISA (n = 3). e Western blot analysis showing the protein levels of IL-6, TNF-α, IL-1β and COX2 in CSE-treated 16HBE cells in each group (n = 3). f A schematic diagram: CSE induced XIST facilitates apoptosis and inflammatory response of 16HBE cells by binding with miR-200c-3p to upregulate EGR3. *p < 0.05, **p < 0.01, ***p < 0.001
Discussion
In our study, we found that CSE treatment promoted apoptosis and inflammation in 16HBE cells, which was consistent with clinical characterization of COPD. The results suggested the successful construction of an in vitro model of COPD. Previously, EGR3 was indicated to exert vital roles in coronary heart disease and systemic lupus erythematosus [9, 25]. In the present study, EGR3 was found to be upregulated in the lung tissues of smokers, especially in smokers with COPD. EGR3 was mainly located in the chronical epithelial cells of airway tissues. EGR3 was reported to promote antigen receptor signaling and control inflammation in adaptive immune responses and homeostasis [10]. In our research, EGR3 silencing suppressed apoptosis and the inflammatory response of CSE-stimulated 16HBE cells. These findings suggested that EGR3 promoted apoptosis and inflammation in CES-treated 16HBE cells.
Next, we aimed to explore the potential upstream mechanism of EGR3 in 16HBE cells. Previously, multiple studies suggested that EGR3 was regulated by several miRNAs at the post-transcriptional level [26,27,28]. In the current study, miR-200c-3p was predicted to be an upstream molecule of EGR3. MiR-200c-3p has been reported to be associated with the development of NSCLC [29]. MiR-200c-3p is a key regulator of bacterial lung infection-induced acute lung injury or acute respiratory distress syndrome [30]. We found that miR-200c-3p was downregulated in the lung tissues of COPD patients. Mechanistically, miR-200c-3p was validated to complementarily bind with the EGR3 3’UTR and negatively regulate EGR3 mRNA and protein levels in 16HBE cells. In addition, miR-200c-3p expression was negatively correlated with EGR3 expression in the lung tissues of smokers. The results suggested that EGR3 was post-transcriptionally regulated by miR-200c-3p in 16HBE cells.
Moreover, miR-200c-3p was reported to interact with XIST in brain microvascular endothelial cells and breast cancer cells [31, 32]. Herein, after prediction by bioinformatics analysis, XIST was found to be a putative lncRNA that binds to miR-200c-3p. Mechanistic experiments revealed that XIST competitively bound with miR-200c-3p and thereby positively modulated EGR3 mRNA and protein levels in 16HBE cells. Furthermore, the XIST expression was negatively correlated with the miR-200c-3p expression and positively correlated with the EGR3 expression in the lung tissues of smokers. A whole transcriptome analysis of human lung tissue identifies XIST as a COPD-associated gene [33]. XIST is a positive regulator of inflammatory response, for example, XIST induces neuroinflammation by targeting the miR-544/STAT3 axis and the miR-137/TNFAIP1 axis in neuropathic pain [34, 35]. XIST mediates the inflammatory response of bovine mammary epithelial cells through the NF-κB/NLRP3 inflammasome pathway [36]. Moreover, XIST was widely reported to promote lung cancer progression [37,38,39]. In the present study, XIST expression was upregulated in the lung tissues of COPD patients. Rescue assays suggested that XIST downregulation inhibited apoptosis and the inflammatory response of CSE-treated 16HBE cells, and these effects were counteracted by EGR3 overexpression. The results suggested that XIST regulated apoptosis and inflammation in CSE-treated 16HBE cells through modulating EGR3 expression.
In conclusion, our study initially confirmed that the XIST and EGR3 is upregulated and miR-200c-3p is downregulated in the airway tissues of smokers and COPD patients. We innovatively put forward that the XIST/miR-200c-3p/EGR3 axis facilitates apoptosis and inflammation of CSE-treated 16HBE cells (Fig. 5f). Therefore, these data will provide a better understanding of XIST-mediated COPD pathogenesis and may provide potential novel insight for the treatment of COPD. However, our investigation on the lncRNA-mediated ceRNA regulatory network involved in COPD progression is still in preliminary phase with some limitations. First, we did not find the correlations between EGR3 and parameters of lung function. Second, sample size limited the confidence of the present study to some degree. Third, the transformed bronchial epithelial cells we used in the present study might distinctly behave from primary bronchial epithelial cells isolated from COPD patients.
Availability of data and materials
The datasets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Eapen MS, Hansbro PM, Larsson-Callerfelt AK, Jolly MK, Myers S, Sharma P, et al. Chronic obstructive pulmonary disease and lung cancer: underlying pathophysiology and new therapeutic modalities. Drugs. 2018;78(16):1717–40.
Tsai MJ, Chang WA, Jian SF, Chang KF, Sheu CC, Kuo PL. Possible mechanisms mediating apoptosis of bronchial epithelial cells in chronic obstructive pulmonary disease—a next-generation sequencing approach. Pathol Res Pract. 2018;214(9):1489–96.
Chapman KR, Mannino DM, Soriano JB, Vermeire PA, Buist AS, Thun MJ, et al. Epidemiology and costs of chronic obstructive pulmonary disease. Eur Respir J. 2006;27(1):188–207.
Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31(6):1334–56.
Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, et al. Chronic obstructive pulmonary disease. Nat Rev Dis Primers. 2015;1:15076.
Brusselle GG, Joos GF, Bracke KR. New insights into the immunology of chronic obstructive pulmonary disease. Lancet (London, England). 2011;378(9795):1015–26.
Boots AW, Gerloff K, Bartholomé R, van Berlo D, Ledermann K, Haenen GR, et al. Neutrophils augment LPS-mediated pro-inflammatory signaling in human lung epithelial cells. Biochim Biophys Acta. 2012;1823(7):1151–62.
Cheng H, Hao S, Liu Y, Pang Y, Ma S, Dong F, et al. Leukemic marrow infiltration reveals a novel role for Egr3 as a potent inhibitor of normal hematopoietic stem cell proliferation. Blood. 2015;126(11):1302–13.
Morita K, Okamura T, Inoue M, Komai T, Teruya S, Iwasaki Y, et al. Egr2 and Egr3 in regulatory T cells cooperatively control systemic autoimmunity through Ltbp3-mediated TGF-β3 production. Proc Natl Acad Sci USA. 2016;113(50):E8131–40.
Li S, Miao T, Sebastian M, Bhullar P, Ghaffari E, Liu M, et al. The transcription factors Egr2 and Egr3 are essential for the control of inflammation and antigen-induced proliferation of B and T cells. Immunity. 2012;37(4):685–96.
Baron VT, Pio R, Jia Z, Mercola D. Early Growth Response 3 regulates genes of inflammation and directly activates IL6 and IL8 expression in prostate cancer. Br J Cancer. 2015;112(4):755–64.
Chien MH, Lee WJ, Yang YC, Li YL, Chen BR, Cheng TY, et al. KSRP suppresses cell invasion and metastasis through miR-23a-mediated EGR3 mRNA degradation in non-small cell lung cancer. Biochim Biophys Acta Gene Regul Mech. 2017;1860(10):1013–24.
Hashimoto N, Matsuzaki A, Okada Y, Imai N, Iwano S, Wakai K, et al. Clinical impact of prevalence and severity of COPD on the decision-making process for therapeutic management of lung cancer patients. BMC Pulm Med. 2014;14:14.
Shen Q, Zheng J, Wang X, Hu W, Jiang Y, Jiang Y. LncRNA SNHG5 regulates cell apoptosis and inflammation by miR-132/PTEN axis in COPD. Biomed Pharmacother Biomed Pharmacother. 2020;126:110016.
Duan R, Niu H, Yu T, Cui H, Yang T, Hao K, et al. Identification and bioinformatic analysis of circular RNA expression in peripheral blood mononuclear cells from patients with chronic obstructive pulmonary disease. Int J Chronic Obstr Pulm Dis. 2020;15:1391–401.
Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505(7483):344–52.
He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5(7):522–31.
Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–33.
Hulina-Tomašković A, Rajković MG, Somborac-Bačura A, Čeri A, Dabelić S, Rumora L. Extracellular Hsp70 modulates the inflammatory response of cigarette smoke extract in NCI-H292 cells. Exp Physiol. 2018;103(12):1704–16.
Song B, Ye L, Wu S, Jing Z. Long non-coding RNA MEG3 regulates CSE-induced apoptosis and inflammation via regulating miR-218 in 16HBE cells. Biochem Biophys Res Commun. 2020;521(2):368–74.
Zhang LH, Wang Z, Li LH, Liu YK, Jin LF, Qi XW, et al. Vestigial like family member 3 is a novel prognostic biomarker for gastric cancer. World J Clin Cases. 2019;7(15):1954–63.
Fan S, Ren Y, Zhang W, Zhang H, Wang C. Long non-coding maternally expressed gene 3 regulates cigarette smoke extract-induced apoptosis, inflammation and cytotoxicity by sponging miR-181a-2-3p in 16HBE cells. Oncol Lett. 2021;21(1):45.
Richter A, O’Donnell RA, Powell RM, Sanders MW, Holgate ST, Djukanović R, et al. Autocrine ligands for the epidermal growth factor receptor mediate interleukin-8 release from bronchial epithelial cells in response to cigarette smoke. Am J Respir Cell Mol Biol. 2002;27(1):85–90.
McKenzie AJ, Hoshino D, Hong NH, Cha DJ, Franklin JL, Coffey RJ, et al. KRAS-MEK signaling controls Ago2 sorting into exosomes. Cell Rep. 2016;15(5):978–87.
Li X, Ma YT, Xie X, Yang YN, Ma X, Zheng YY, et al. Association of Egr3 genetic polymorphisms and coronary artery disease in the Uygur and Han of China. Lipids Health Dis. 2014;13:84.
Li X, Yuan M, Song L, Wang Y. Silencing of microRNA-210 inhibits the progression of liver cancer and hepatitis B virus-associated liver cancer via targeting EGR3. BMC Med Genet. 2020;21(1):48.
Wang ZD, Qu FY, Chen YY, Ran ZS, Liu HY, Zhang HD. Involvement of microRNA-718, a new regulator of EGR3, in regulation of malignant phenotype of HCC cells. J Zhejiang Univ Sci B. 2017;18(1):27–36.
Zhang P, Yang X, Wang L, Zhang D, Luo Q, Wang B. Overexpressing miR-335 inhibits DU145 cell proliferation by targeting early growth response 3 in prostate cancer. Int J Oncol. 2019;54(6):1981–94.
Wang HY, Liu YN, Wu SG, Hsu CL, Chang TH, Tsai MF, et al. MiR-200c-3p suppression is associated with development of acquired resistance to epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in EGFR mutant non-small cell lung cancer via a mediating epithelial-to-mesenchymal transition (EMT) process. Cancer Biomark Sect A Dis Mark. 2020;28(3):351–63.
Liu Q, Du J, Yu X, Xu J, Huang F, Li X, et al. miRNA-200c-3p is crucial in acute respiratory distress syndrome. Cell Discov. 2017;3:17021.
Wang S, Han X, Mao Z, Xin Y, Maharjan S, Zhang B. MALAT1 lncRNA induces autophagy and protects brain microvascular endothelial cells against oxygen–glucose deprivation by binding to miR-200c-3p and upregulating SIRT1 expression. Neuroscience. 2019;397:116–26.
Zhang M, Wang F, Xiang Z, Huang T, Zhou WB. LncRNA XIST promotes chemoresistance of breast cancer cells to doxorubicin by sponging miR-200c-3p to upregulate ANLN. Clin Exp Pharmacol Physiol. 2020;47(8):1464–72.
Zhu Y, Zhou A, Li Q. Whole transcriptome analyis of human lung tissue to identify COPD-associated genes. Genomics. 2020;112(5):3135–41.
Jin H, Du XJ, Zhao Y, Xia DL. XIST/miR-544 axis induces neuropathic pain by activating STAT3 in a rat model. J Cell Physiol. 2018;233(8):5847–55.
Zhao Y, Li S, Xia N, Shi Y, Zhao CM. Effects of XIST/miR-137 axis on neuropathic pain by targeting TNFAIP1 in a rat model. J Cell Physiol. 2018;233(5):4307–16.
Ma M, Pei Y, Wang X, Feng J, Zhang Y, Gao MQ. LncRNA XIST mediates bovine mammary epithelial cell inflammatory response via NF-κB/NLRP3 inflammasome pathway. Cell Prolif. 2019;52(1):e12525.
Liu J, Yao L, Zhang M, Jiang J, Yang M, Wang Y. Downregulation of LncRNA-XIST inhibited development of non-small cell lung cancer by activating miR-335/SOD2/ROS signal pathway mediated pyroptotic cell death. Aging. 2019;11(18):7830–46.
Wang X, Zhang G, Cheng Z, Dai L, Jia L, Jing X, et al. Knockdown of LncRNA-XIST suppresses proliferation and TGF-β1-induced EMT in NSCLC through the Notch-1 pathway by regulation of miR-137. Genet Test Mol Biomark. 2018;22(6):333–42.
Jiang H, Zhang H, Hu X, Li W. Knockdown of long non-coding RNA XIST inhibits cell viability and invasion by regulating miR-137/PXN axis in non-small cell lung cancer. Int J Biol Macromol. 2018;111:623–31.
Acknowledgements
Thank you for all lab members involved in this study.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
PC conceived and designed the experiments. PC, PJ, JC, YY, and XG carried out the experiments. PC and PJ analyzed the data. PC drafted the manuscript. All authors agreed to be accountable for all aspects of the work. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from all participants. The Ethics Committee of Tianjin First Central Hospital approved our study. The research has been carried out in accordance with the World Medical Association Declaration of Helsinki.
Consent for publication
Not applicable.
Competing interests
The authors declare that there exists no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The related sequences in this research.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, P., Jiang, P., Chen, J. et al. XIST promotes apoptosis and the inflammatory response in CSE-stimulated cells via the miR-200c-3p/EGR3 axis. BMC Pulm Med 21, 215 (2021). https://doi.org/10.1186/s12890-021-01582-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-021-01582-8