
Abstract
Background
This study assessed the effects of ursolic acid (UA) on airway-vessel remodeling and muscle atrophy in cigarette smoke (CS)-induced emphysema rats and investigated potential underlying mechanisms.
Methods
Emphysema was induced in a rat model with 3 months of CS exposure. Histology and immunohistochemistry (IHC) stains were used to assess airway-vessel remodeling and muscle atrophy-associated changes. Levels of cleaved-caspase3, 8-OHdG, and S100A4 were measured in airways and associated vessels to evaluate cell apoptosis, oxidant stress, epithelial-to-mesenchymal transition (EMT), and endothelial-to-mesenchymal transition (EndMT)-associated factors. Western blot and/or IHC analyses were performed to measure transforming growth factor-beta 1(TGF-β1)/Smad2.3, alpha-smooth muscle actin (α-SMA), and insulin-like growth factor 1 (IGF1) expression. We also gave cultured HBE and HUVEC cells Cigarette Smoke Extract (CSE) administration and UA intervention. Using Western blot method to measure TGF-β1/Smad2.3, α-SMA, S100A4, and IGF1 molecules expression.
Results
UA decreased oxidant stress and cell apoptosis in airway and accompanying vascular walls of cigarette smoke-induced emphysema model rats. UA alleviated EMT, EndMT, changes associated with airway-vessel remodeling and muscle atrophy. The UA effects were associated with IGF1 and TGF-β1/Smad2.3 pathways.
Conclusions
UA reduced EMT, EndMT, airway-vessel remodeling, and musculi soleus atrophy in CS-induced emphysema model rats at least partly through IGF1 and TGF-β1/Smad2.3 signaling pathways.
Similar content being viewed by others


Background
Chronic obstructive pulmonary disease (COPD) is a systemic disease characterized by persistent respiratory symptoms and airway limitation. It has high prevalence and associated mortality, with a prevalence among people 40 years of age or older of 10.1% worldwide and 13.7% in China [1, 2] and was estimated to be the third leading cause of death worldwide in 2030 [3]. The disease gives rise to enormous social and economic burdens [4, 5]. Nearly 90% of COPD cases are caused by Cigarette Smoke (CS) [6, 7]. Major pathological manifestations of COPD include chronic airway inflammation, airway-vessel remodeling [8], and emphysema [9]. Muscle atrophy is an important complication of COPD [10]. Although they are significant indicators of poor prognosis [8, 11,12,13], there are currently no effective interventions for airway-vessel remodeling and muscle consumption in COPD.
Airway-vessel remodeling is the main contributor to pulmonary dysfunction in COPD [14]. Potential mechanisms contributing to the airway-vessel remodeling of COPD include proliferation of airway epithelial cells, vascular endothelial cells, airway and vessel smooth muscle cells, fibroblast-myofibroblast transformation, epithelial-to-mesenchymal transition (EMT), and endothelial-to-mesenchymal transition (EndMT) [15,16,17]. Beyond the well-known airway remodeling processes associated with COPD [18], there has been a growing interest in vessel remodeling in COPD [19,20,21,22], for which the mechanism is not yet clear. The 2018 GOLD guide suggested that pulmonary microvascular blood flow was abnormal in smokers with even mild COPD. Meanwhile, patients with moderate or severe COPD often show pronounced pulmonary vascular remodeling, leading to pulmonary hypertension and pulmonary heart disease, which are directly related to patient prognosis. Patients with combined pulmonary fibrosis and emphysema, which are on the rise, are at increased risk of pulmonary hypertension and have a worse prognosis than patients with emphysema only, further indicating that vessel remodeling affects COPD progression [23].
CS activated cell oxidant stress and apoptosis can promote Transforming growth factor-beta 1(TGF-β1) secretion [24,25,26,27]. The TGF-β1/Smads signaling pathways are thought to mediate CS-induced airway-vessel remodeling in COPD [8, 28,29,30,31]. TGF-β1 is a multi-functional cytokine that regulates angiogenesis, extracellular matrix deposition, and fibroblast/myofibroblast trans-differentiation [29, 32,33,34]. Among its downstream pathways, TGF-β1/Smad2.3 signaling is strongly implicated in EMT and EndMT, which play key roles in COPD-associated airway-vessel remodeling [15, 28, 30].
Sarcopenia is an important complication of COPD and an indicator of poor prognosis of COPD patients [12, 35]. Exercise and glucocorticoids stimulate muscle recovery with variable efficacy, depending on the patient’s clinical condition and medical treatment [36]. Insulin-like growth factor 1(IGF1) is thought to play a key role in bronchial epithelial and muscle cell regeneration in COPD patients [36,37,38,39]. Thus, IGF1 intervention may contribute to treat COPD through effects on airway-vessel remodeling and muscle atrophy.
Ursolic acid (UA), a pentacyclic triterpenoid compound exits in many plants. It has anti-oxidant [40, 41], anti-inflammatory [42], anti-tumor [43], anti-apoptotic [44], and anti-fibrotic effects [45], all of which could support COPD treatment. In our previous experiment, we found UA intervention could alleviate CS induced emphysema in rats [46]. Researchers reported previously that UA is an antagonist of TGF-β1 [47], but whether UA exerts its effect through TGF-β1/Smads pathways remained unknown, especially in the context of COPD. UA was also reported to alleviate muscle consumption through the IGF1 pathway in an animal model of chronic kidney disease [48]. However, whether UA can alleviate CS-induced airway remodeling and muscle consumption in emphysema rats, and whether UA exerts its effects through TGF-β1/Smads and IGF1 pathways, remains to be established.
Therefore, we used CS induced rat emphysema model to assessed the effect of UA on EMT, EndMT, airway-vessel remodeling and muscle consumption and discuss the underlying mechanisms through TGF-β1/Smads pathways and IGF1 molecule. This study offered a new ademption for the treatment of clinical COPD patients.
Methods
Compounds and reagents
Antibodies against TGF-β1, 8-OHdG, α-SMA, S100A4, and IGF1 were obtained from Abcam Biotechnology Company (Cambridge, UK). Antibodies against Smad2, p-Smad2, Smad3, p-Smad3, and cleaved-caspase3 were purchased from Cell Signaling Technology (Denver, CO). UA was purchased from Wanxiang Hengyuan Biotechnology (Tianjin, China). Masson and Alcian blue-periodic acid Schiff (AB-PAS) kits were obtained from Nanjing Jiancheng biological engineering research institute (Nanjing, China). BCA kit was obtained from Pierce (Thermo-Scientific, Rockford, IL, USA), ECL chemiluminescence kit from Applygen (Beijing, China).
Animals
Six-week-old male Wistar rats, weighing between 150 and 250 g, were bought from Chansheng Biotechnology Company (Liaoning, China). After two weeks of adaptation time, rats were randomized into one of five treatment groups (N = 10 each): Sham, CS, UA10, UA20, and UA40. UA rats were administrated 10 mg/kg, 20 mg/kg or 40 mg/kg body weight UA via gavage thirty minutes before the first CS exposure every day. Sham and CS rats were given vehicle instead. CS and UA rats were exposed to smoke of 16 filters removed 1R3F cigarettes for 30 min, two times a day, 6 days a week, for 3 months. Rats from the same group were placed five at a time into a glass chamber measuring 0.8 m × 0.6 m × 0.6 m, with a 2 cm × 2 cm spiracle on the top of the box. The time interval between the two exposures each day was 4 to 6 h. Rats in the Sham group were exposed to normal air. [46] The Animal Care and Use Ethics Committee of China Medical University approved this study.
Cells culture and interventions
Human bronchial epithelial cells (HBEs) and human umbilical vein endothelial cells (HUVECs) were purchased from Peking University Cancer Institute (Beijing, China). Cells were cultured in RPMI-1640 culture medium (Hyclone, UT, USA) containing 10% fetal bovine serum (Hyclone, UT) in a 5% CO2 humidified cell incubator (Thermo Fisher Scientific, Inc., USA) at 37 °C. We treated 1 × 106 cells with 10 μM/L UA 2 h prior to 1% cigarette smoke extract (CSE) [49] intervention. The concentrations we used were according to the CCK8 cytotoxicity testing (Dojido, Japan).
Pathological materials
The left lungs were inflated and fixed using 4% phosphate- buffered formaldehyde (pH 7.40) at 25cmH2O pressure for 24 h. The musculi soleus muscles were fixed using 10% formaldehyde for 24 h. Then lungs and musculi soleus muscles embedded with paraffin. The paraffin-embedded sections were used for histopathological examination. Right lung tissues were frozen in liquid nitrogen for 5 min before storing at − 80 °C.
Histopathology
We used hematoxylin and eosin (HE) staining to observe pathological changes to pulmonary airways and vessels. We measured and compared mean thickness of the airways and associated vessels in lung tissues. We observed and compared the pathological changes of musculi soleus using HE staining. To evaluate bronchial and vascular wall thickness, four sections that did not include cartilage but did include intact bronchial tracheal transections and concomitant vessels were randomly selected. For all bronchial sections, the ratio of minimum diameter to maximum diameter was> 0.5. Image Pro Plus 5.0 image analysis software (Media Cybernetics company, Maryland, American) was used to measure airway and vascular basement membrane perimeter (Pbm), airway wall area (total wall area [Wat]) and vascular wall area (total vascular area [Vat]). Wat/Pbm and Vat/Pbm values were calculated for each trachea and associated vessel, and the average value was used to compared airway and vascular wall thicknesses among groups.
Masson staining
We used Masson staining to measure collagen deposition around the airways and vessels. Paraffin sections were dewaxed to water, then stained according to the Masson staining kit instructions (Nanjing Jiancheng Biotechnology, Nanjing, China). Areas of collagen deposition around airways and vessels were compared using the index wall area of collagen deposition (Wac)/Wat and vascular area of collagen deposition (Vac)/Vat measured using Image Pro Plus 5.0 image analysis software.
AB-PAS staining
We used AB-PAS staining to count mucus-producing cells surrounding the airway. Paraffin sections were dewaxed to water, then stained according to the AB-PAS staining kit instructions (Nanjing Jiancheng Biotechnology, China).
Immunohistochemistry (IHC)
Briefly, paraffin embedded tissues cut into 4-μm thick sections, and dewaxed, rehydrated. The slices were treated with H2O2 in methanol to inhibit endogenous peroxidase activity. Then antigen retrieval was performed using a microwave and 10-mM citrate buffer, pH 6. Slices were incubated with anti-8-OHdG, anti-cleaved caspase-3, anti-α-SMA, anti-TGF-β1, anti-p-Smad2, and anti-S100A4 antibodies overnight with the concentration of 1:500 at 4 °C. After washing, secondary antibodies were incubated room temperature for 1 h using the concentration of 1:1000. Then incubation with 3,3′-diaminobenzidine (DAB) and DAB Enhancer. One horizon in each quadrant of each section was assessed. Relative expression was compared using relative integrated optical density (IOD) surrounding the airway (IOD/Wat) and vessel (IOD/Vat), as measured using IPP 5.0 software. [46]
Western blot analysis
Lung tissues, HBE cells and HUVEC cells lysates were prepared. Briefly, tissues and cells were lysed in an ice-cold lysis buffer (Roche Applied Science, Indianapolis, IN). Samples were then homogenized for 15 s at 4 °C, 4–5 times. Cell lysates were centrifuged at 12,000×g for 30 min at 4 °C to remove cellular debris. Protein concentration was determined using a BCA protein assay kit. Equal amounts of protein (20–60 μg) were separated on 8–10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and then transferred to PVDF membranes (Merck Millipore, Darmstadt, Germany), blocked and incubated with diluted primary antibodies overnight at 4 °C refrigerator. Blots were stripped and re-probed with anti-GAPDH antibody to demonstrate equal loading. Incubated with secondary antibody, the chemiluminescent signal was detected using the Super Enhanced Chemiluminescence Kit (Bio-Rad Laboratories, Shanghai, China). Band density was quantified using Quantity One software (Bio-Rad Laboratories, Shanghai, China). [46]
Statistical analysis
SPSS13.0 software and Graph Pad Prism 5.0 software were used for statistical analysis. Kolmogorov Smirnov and Shapiro tests were used to assess normality, and all the data fit a Gaussian distribution. Data are presented as Mean ± Standard Deviation (SD). One-way analyses of variance (ANOVAs) were used to compare differences among groups. p < 0.05 was considerate to with statistical difference.
Results
UA decreased musculi soleus atrophy in CS-induced emphysema rats
We had 10 rats in each group at the beginning of the experiment, but 2 rats in the CS group died during CS exposure. The remaining 48 rats survived and were used in subsequent experiments. Musculi soleus weights were significantly reduced in the CS group, relative to the Sham group, and 40 mg/kg body weight UA administration significantly attenuated this muscle mass loss. The same trend was not observed for extensor digitorum longus (Fig. 1A). Examination of HE stained sections revealed marked muscle atrophy with characteristic muscle cell crinkle, cell vacuolation, and structural disorder in the musculi soleus of CS-induced emphysema model rats, relative to the muscles of Sham rats. In UA groups, musculi soleus atrophy related changes such as muscle cell crinkle, vacuolation, muscle fiber disorder, and gap dilation were all improved in a concentration dependent manner. Myoarchitecture in UA20 and UA40 groups were similar to that of the Sham group (Fig. 1B).

UA attenuate CS induced muscle atrophy in emphysema model rats. A Weights of musculi soleus and extensor digitorum longus. 40 mg/kg body weight UA administration attenuated CS induced musculi soleus loss. #p < 0.05 vs. Sham; *p < 0.05 vs. CS. B Morphological changes of musculi soleus. HE staining of musculi soleus showed muscle atrophy associate changes in CS, and these changes were alleviated by UA intervention, especially in UA20 and UA40. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). UA: ursolic acid; CS: cigarette smoke (group); UA10: 10 mg/kg body weight UA administration group; UA20: 20 mg/kg body weight UA administration group; UA40: 40 mg/kg body weight UA administration group
UA alleviated airway-vessel remodeling in CS-induced emphysema rat lungs
HE stained sections revealed that the airways and associated vessels of the emphysema rats underwent airway-vessel remodeling, UA administration alleviated CS induced airway and vessel remodeling. The pathology changes observed in CS rats included proliferation and exfoliation of airway epithelial cells, airway basement membrane thickening, collagen deposition, airway contracture, infiltration of inflammation cells around airways, and vessel thickening. UA administration at all three concentrations alleviated these changes (Fig. 2A). We used airway and vessel thickness to quantify changes associated with airway-vessel remodeling. Compared with Sham group, the thickness of airways and accompanying vessels were remarkably increased with CS, and UA administration alleviated these changes at all three concentrations tested (Fig. 2B).

UA alleviate airway-vessel remodeling in emphysema model rats. A HE staining of lung sections showed that UA administration reduced the airway-vessel remodeling associated changes in CS-induced emphysema rats. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). B Airway thickness (Wat/Pbm: μm2/μm) and vascular thickness (Vat/Pbm: μm2/μm). ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. C Masson staining of lung sections showed that UA alleviated collagen deposition in tissue surrounding airways and accompanying vessels in lungs of emphysema rats. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). D Collagen deposition in areas surrounding airways (Wac/Wat: μm2/μm2) and vessels (Vac/Vat: μm2/μm2). ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. E PAS staining of lung sections showed that UA alleviated mucus secretion cell expression around airways in lungs of emphysema rats. Pink arrows show goblet cells. Black arrow shows mucous gland near the major airway. UA: ursolic acid; CS: cigarette smoke (group); UA10: 10 mg/kg body weight ursolic acid administration group; UA20: 20 mg/kg body weight ursolic acid administration group; UA40: 40 mg/kg body weight ursolic acid administration group; Pbm: airway or vessel basement membrane perimeter; Wat: airway wall area; Vat: vascular wall area; Wac: wall area of collagen deposition; Vac: vascular area of collagen deposition
Effects of UA on collagen deposition around airways and vessels caused by CS exposure
Masson staining showed that blue-dyed collagen deposition areas were significantly increased in airway and accompanying vessel walls of CS rats, compared with those in the Sham rats. Blue-dyed areas around airways and accompanying vessels were reduced significantly by all doses of UA administered (Fig. 2C). We used blue-stained areas in airway or vessels divided by the total areas of airway or vessels to compare the relative extent of collagen deposition. Proportion of collagen deposition areas to total airway or vessel areas are increased significantly in CS group compared with Sham. UA administration reduced the proportion of collagen deposition around airways and accompanying vessels (Fig. 2D).
Effects of UA on airway mucus secretion cell expression caused by CS exposure
We used AB-PAS staining to assess mucus secreting cell expression surrounding the airways. Many large blue-dyed goblet cells were present in the epithelial layer in the airways of CS rats compared with Shams. UA decreased the size and number of goblet cells in epithelial layer of CS rats. We also noted metaplasia of large blue-dyed mucous glands in the large airway in the CS group (Fig. 2E).
Effects of UA on airway and vessel smooth muscle cell proliferation caused by CS exposure
To investigate smooth muscle cell proliferation, fibroblast to myofibroblast differentiation, EMT, and EndMT, we examined α-SMA expression in the area surrounding the airways and accompanying vessels using IHC. α-SMA expression around airways and vessels were significantly increased in the CS group compared with the Sham group, especially in the epithelial and endothelial layers. UA decreased α-SMA expression in the area surrounding airways and vessels, particularly in the epithelial and endothelial layers, at each dose of UA (Fig. 3A). The percentage of α-SMA staining areas per airway or vessel areas were significantly increased in CS rats compared with Sham rats, but UA administration decreased the percent of α-SMA staining area at all doses (Fig. 3B).

UA alleviate oxidant stress and cell apoptosis in airways and associated vessels, attenuated EMT/EndMT in CS-induced emphysema rats. A IHC staining of lung sections showed that UA reduced α-SMA expression in airways and vessels in emphysema rat lung, especially in the epithelial and endothelial layers. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). B IHC IOD of α-SMA expression in airways and vessels. IOD/Wat: /μm2 and IOD/Vat: /μm2 data are shown. #p < 0.05 ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. C IHC staining of lung sections showed that UA reduced S100A4 expression in airway and vessel, especially in the epithelial and endothelial layers. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). D IHC IOD of S100A4 expression in airways and vessels. IOD/Wat: /μm2 and IOD/Vat: /μm2 data are shown. #p < 0.05, ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. E IHC staining of lung sections showed that UA reduced 8-OHdG expression in airways and accompanying vessels in emphysema rat lungs, especially in the epithelial and endothelial layers. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). F IHC IOD of 8-OHdG expression in airways and vessels. IOD/Wat: /μm2 and IOD/Vat: /μm2 data are shown. #p < 0.05, ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. G IHC staining of lung sections showed that UA reduced cleaved-caspase3 expression in airways and accompanying vessels in emphysema rat lungs, especially in the epithelial and endothelial layers. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). H IHC IOD of cleaved-caspase3 expression in airways and vessels. IOD/Wat: /μm2 and IOD/Vat: /μm2 data are shown. ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. UA: ursolic acid; CS: cigarette smoke (group); UA10: 10 mg/kg body weight UA administration group; UA20: 20 mg/kg body weight UA administration group; UA40: 40 mg/kg body weight UA administration; Pbm: airway or vessel basement membrane perimeter; Wat: airway wall area; Vat: vascular wall area; IHC: immunohistochemistry; IOD: integral optical density
Effects of UA on EMT, EndMT in airways and accompanying vessels caused by CS exposure
We used S100A4 expression to assess EMT and EndMT around airways and vessels. IHC analyses showed that S100A4 expression surrounding airways and accompanying vessels was significantly increased in the CS group compared with the Sham group. This increase was suppressed by UA, especially in the epithelial and endothelial layers (Fig. 3C). The percentage of S100A4 staining areas per airway or vessel areas were significantly increased in CS rats compared with Sham rats, and UA administration reversed this effect at all doses (Fig. 3D).
Effects of UA on oxidant stress and cell apoptosis around airways and vessels caused by CS exposure
We used 8-OHdG and cleaved-caspase3 expressions to assess cell apoptosis around airways and vessels. IHC analyses showed that 8-OHdG and cleaved-caspase3 expression surrounding airways and vessels was significantly increased in the CS group compared with the Sham group. This increase was suppressed by UA, particularly in the epithelial and endothelial layers (Fig. 3E, G). The IOD/area values are consistent with the results above (Fig. 3F, H).
Effects of UA on TGF-β1/Smad2.3 pathway and IGF1 expression caused by CS exposure
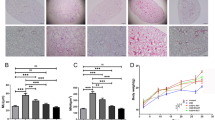
IHC analysis further showed markedly upregulated TGF-β1 expression in airways and accompanying vessels, while in UA groups, TGF-β1 expression was downregulated in all of the UA groups, especially in the epithelial and endothelial layers (Fig. 4A). These IOD/area values are consistent with the results above (Fig. 4B). Western blot analysis of TGF-β1, p-Smad2, p-Smad3, Smad2, Smad3, α-SMA, and IGF1 expression showed that the activated forms of TGF-β1, p-Smad3, and α-SMA were upregulated in rat lung in the CS group compared with the Sham group. In the UA groups, active forms expression of TGF-β1, p-Smad3, and α-SMA was down-regulated. IGF1 expression was down-regulated in the CS group compared with the Sham group. UA administration upregulated IGF1 expression in rat lung (Fig. 4C and D). Contrary to expectations, the p-Smad2 expression seemed to be down-regulated in the CS group but upregulated in the UA groups. Thus, we used IHC to assess p-smad2 expression around airways and accompanying vessels. It revealed markedly upregulated p-Smad2 expression in airways and accompanying vessels, but not pulmonary parenchyma regions, downregulated p-Smad2 expression in airways and accompanying vessels after UA treatment, which is inconsistent with the IOD results (Fig. 4E and F).

UA intervention alleviate TGF-β1/smad2.3 pathway molecules expression in airways and accompanying vessels of emphysema rats. A IHC staining of lung sections showed that UA reduced TGF-β1 expression in tissue surrounding airways and vessels in rat lungs. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). B IHC IOD of TGF-β1 expression in airways and vessels. IOD/Wat and IOD/Vat: /μm2 data are shown. ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. C Western blot analysis of TGF-β1/Smad2.3 pathway molecules and IGF1 expression in the lungs of emphysema rats. D Western blot IOD of TGF-β1/Smad2.3 pathway molecules and IGF expression in lungs. #p < 0.05 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. E IHC staining of lung sections showed that UA reduced p-Smad2 expression in tissue surrounding airways and vessels in rat lungs. Sham (a), CS (b), UA10 (c), UA20 (d), and UA40 (e). F IHC IOD of p-Smad2 expression in airways and vessels. IOD/Wat and IOD/Vat: /μm2 data are shown. ##p < 0.01 vs. Sham; *p < 0.05, **p < 0.01 vs. CS. UA: ursolic acid; CS: cigarette smoke (group); UA10: 10 mg/kg body weight UA administration group; UA20: 20 mg/kg body weight UA administration group; UA40: 40 mg/kg body weight UA administration; Pbm: airway or vessel basement membrane perimeter; Wat: airway wall area; Vat: vascular wall area; IHC: immunohistochemistry; IOD: integral optical density
Effects of UA on TGF-β1/Smad2.3 pathway molecules expression in HBE and HUVEC cells after CSE administration
Western blot analysis showed upregulated expression of activated forms of TGF-β1, p-Smad2, p-Smad3, and S100A4 in HBEs and HUVECs after CSE administration compared with vehicle control cells, that could be attenuated by UA treatment. CSE administration downregulated IGF1 expression in HBE cells, UA alleviated the IGF1 down-regulation (Fig. 5A-D).

UA alleviated CSE-induced TGF-β1/Smad2.3 pathway proteins and S100A4 expression in HBEs and HUVECs in vitro. A Western blot analyses showed UA attenuated CSE-induced TGF-β1/Smad2.3 pathway protein and S100A4 expression in HBEs. B IOD of TGF-β1/smad2.3 pathway, IGF1, and S100A4 expression in HBEs
#p < 0.05 vs. Sham; *p < 0.05 vs. CSE. C Western blot analyses showed that UA alleviated CSE-induced TGF-β1/Smad2.3 and S100A4 protein expression in HUVECs. D IOD of TGF-β1/Smad2.3 pathway proteins and S100A4 expression in HUVECs. #p < 0.05 vs. Sham; *p < 0.05 vs. CSE. UA: ursolic acid; CSE: cigarette smoke extract (group); HBE: Human Bronchial Epithelial cells; HUVEC: Human Umbilical Vein Endothelial Cells; IOD: integral optical density.
Discussion
The present study showed that UA treatment alleviated EMT, EndMT, airway-vessel remodeling, and muscle atrophy-associated lesions in a rat model of CS-induced emphysema. We further demonstrated that UA exerts its effects through mechanisms that involve upregulation of IGF-1 and inhibition of the TGF-β1/Smad2,3 pathway.
Similar to EMT, of which it is a special type, EndMT refers to the process of under-stimulated endothelial cells taking on a mesenchymal cell phenotype. EndMT is characterized by decreased cellular connectivity, reduced expression of endothelial markers (e.g., CD31 and cadherin), increased expression of mesenchymal markers (e.g., α-SMA and vimentin), and signs of cellular invasion and migration. Recent observations of upregulated expression of S100A4 in the vascular walls of COPD patients suggest that EndMT may be involved in COPD pulmonary vascular remodeling [16, 17], though the mechanism underlying these changes is unclear.
We found in this study, 3 months of CS exposure induced significant airway-vascular remodeling in rat lungs. Pathological manifestations included increased thickness of the airways and accompanying vessels, as well as collagen deposition in these areas. Remodeling increased the size and quantity of goblet cells in the epithelial layer of the airway, as well as mucinous gland metaplasia in areas surrounding the central airway. Remodeling also involved increased expression of α-SMA and S100A4 in airways and accompanying vessels. Expression of S100A4 is specific index for EMT and EndMT.
3 months of CS exposure also increased TGF-β1 expression in airway and vessel walls as well as whole lung of rats, and its downstream p-Smad3 expression. However, western blot analyses showed decreased p-Smad2 expression in whole lung of rats, contrasting with previous findings [50]. We sought to explain this discrepancy.
In a previous study of Smad2 activation in the lung tissues of COPD patients, Lepparanta and colleagues found reduced Smad2 activation in alveoli and increased Smad2 activation in thickened bronchial tissues. Down-regulation of p-Smad2 expression in emphysema rat lungs may due to imbalanced expression of Smad2 in pulmonary parenchyma and airway-vessels [51]. Our IHC analysis of p-Smad2 expression in airways and vessels showed upregulation of p-Smad2 in airway and vessel walls of CS-induced emphysema model rats. We also observed higher levels of TGF-β1/Smad2,3 pathway constituents and increased S100A4 expression in CSE-exposed HBEs and HUVECs.
UA, a compound that comprises three terpenoids found in plants, has a wide range of effects, which may inhibit the occurrence and development of COPD. Previously, we found that UA administration significantly alleviated body weight loss, oxidative stress, and cell apoptosis in lung tissue of CS induced emphysema rats. UA exerted its effects through the unfolded protein response (UPR) PERK and Nrf2 pathways [46]. We also found previously that IRE1 pathway, but not ATF6 pathway, signaling was upregulated this model. In this study, we found that UA alleviated EMT, EndMT, airway-vessel remodeling, and muscular atrophy in the same model, and that it does so partly through TGF-β/Smad2.3 and IGF-1 signaling pathways. These results suggest that UA could exert dual effects in rats with CS-induced emphysema.
The UPR of endoplasmic reticulum stress (ERS) has been described involving in EMT in other disease processes [52,53,54,55]. It is not yet known whether a similar activation of the UPR occurs during EMT of airway and alveolar epithelial cells in COPD. Recently, Liang and colleagues proposed that ERS induced by advanced oxidation protein products may be involved in glomerular endothelial cell EndMT, leading to the development of diabetic nephropathy [56]. Meanwhile, Ying and colleagues proposed that ERS-induced EndMT may occur through the Src pathway in HUVECs [57]. It remains to be established whether activation of the unfolded protein response plays a role in EMT/EndMT during COPD-associated airway-vessel remodeling, and if so, which pathways are most critical. Furthermore, it will be of interest to determine whether UA treatments that alleviate airway-vessel remodeling affect the unfolded protein response in association with endoplasmic reticulum stress.
However, our findings are insufficient to identify the exact mechanism underlying the effects of UA on CS-induced airway-vessel remodeling. The nature of the link between EndMT and COPD has yet to be clarified [16, 17]. These observations are important and warrant further studies.
Conclusion
Adding to our previous study showing that UA can alleviate CS-induced emphysema in rats via attenuation of oxidative stress and cell apoptosis, here we show that UA can also alleviate CS-induced EMT, EndMT, airway-vessel remodeling, and muscle atrophy. As a compound that occurs naturally in plants, and has already been used for clinical trials in solid tumors, UA offers much promise as an intervention for the pathogenesis, symptoms, and complications of COPD.
Abbreviations
- EMT:
-
epithelial-to-mesenchymal transition
- EndMT:
-
endothelial-to-mesenchymal transition
- IGF1:
-
insulin-like growth factor 1
- Pbm:
-
basement membrane perimeter
- TGF-β1:
-
transforming growth factor-beta 1
- UA:
-
ursolic acid
- Vac:
-
vascular area of collagen deposition
- Vat:
-
total vascular area
- Wac:
-
wall area of collagen deposition
- Wat:
-
total wall area
References
Buist AS, McBurnie MA, Vollmer WM, Gillespie S, Burney P, Mannino DM, Menezes AM, Sullivan SD, Lee TA, Weiss KB, et al. International variation in the prevalence of COPD (the BOLD study): a population-based prevalence study. Lancet. 2007;370(9589):741–50.
Wang C, Xu J, Yang L, Xu Y, Zhang X, Bai C, Kang J, Ran P, Shen H, Wen F, et al. Prevalence and risk factors of chronic obstructive pulmonary disease in China (the China pulmonary health [CPH] study): a national cross-sectional study. Lancet. 2018;391(10131):1706–17.
T O, K N, M T, S S. T H: analysis of the factors related to mortality in chronic obstructive pulmonary disease: role of exercise capacity and health status. Am J Respir Crit Care Med. 2003;167(4):544–9.
Zhong N, Wang C, Yao W, Chen P, Kang J, Huang S, Chen B, Wang C, Ni D, Zhou Y, et al. Prevalence of chronic obstructive pulmonary disease in China: a large, population-based survey. Am J Respir Crit Care Med. 2007;176(8):753–60.
Oga T, Nishimura K, Tsukino M, Sato S, Hajiro T. Analysis of the factors related to mortality in chronic obstructive pulmonary disease: role of exercise capacity and health status. Am J Respir Crit Care Med. 2003;167(4):544–9.
McMillan DH, Baglole CJ, Thatcher TH, Maggirwar S, Sime PJ, Phipps RP. Lung-targeted overexpression of the NF-kappaB member RelB inhibits cigarette smoke-induced inflammation. Am J Pathol. 2011;179(1):125–33.
Gould NS, Min E, Gauthier S, Chu HW, Martin R, Day BJ. Aging adversely affects the cigarette smoke-induced glutathione adaptive response in the lung. Am J Respir Crit Care Med. 2010;182(9):1114–22.
Soltani A, Walters EH, Reid DW, Shukla SD, Nowrin K, Ward C, Muller HK, Sohal SS. Inhaled corticosteroid normalizes some but not all airway vascular remodeling in COPD. International journal of chronic obstructive pulmonary disease. 2016;11:2359–67.
Baddini-Martinez J, de Padua AI. Chronic obstructive pulmonary disease: time to discuss new concepts. Lancet. 2016;388(10061):2740–1.
Barreiro E, Gea J. Molecular and biological pathways of skeletal muscle dysfunction in chronic obstructive pulmonary disease. Chronic respiratory disease. 2016;13(3):297–311.
Blanco I, Piccari L, Barbera JA. Pulmonary vasculature in COPD: the silent component. Respirology (Carlton, Vic). 2016;21(6):984–94.
Costa TM, Costa FM, Moreira CA, Rabelo LM, Boguszewski CL, Borba VZ. Sarcopenia in COPD: relationship with COPD severity and prognosis. Jornal brasileiro de pneumologia : publicacao oficial da Sociedade Brasileira de Pneumologia e Tisilogia. 2015;41(5):415–21.
Pera T, Zuidhof AB, Smit M, Menzen MH, Klein T, Flik G, Zaagsma J, Meurs H, Maarsingh H. Arginase inhibition prevents inflammation and remodeling in a Guinea pig model of chronic obstructive pulmonary disease. J Pharmacol Exp Ther. 2014;349(2):229–38.
Alagappan VK, de Boer WI, Misra VK, Mooi WJ, Sharma HS. Angiogenesis and vascular remodeling in chronic airway diseases. Cell Biochem Biophys. 2013;67(2):219–34.
Sohal SS. Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir Investig. 2017;55(2):104–13.
Sohal S. Epithelial and endothelial cell plasticity in chronic obstructive pulmonary disease (COPD). Respir Investig. 2017;55(2):104–13.
Sohal S. Endothelial to mesenchymal transition (EndMT): an active process in chronic obstructive pulmonary disease (COPD)? Respir Res. 2016;17:20.
Hirota N, Martin J. Mechanisms of airway remodeling. Chest. 2013;144(3):1026–32.
Takayanagi S, Kawata N, Tada Y, Ikari J, Matsuura Y, Matsuoka S, Matsushita S, Yanagawa N, Kasahara Y, Tatsumi K. Longitudinal changes in structural abnormalities using MDCT in COPD: do the CT measurements of airway wall thickness and small pulmonary vessels change in parallel with emphysematous progression? Int J Chron Obstruct Pulmon Dis. 2017;12:551–60.
Fathy E, Shafiek H, Morsi T, El Sabaa B, Elnekidy A, Elhoffy M, Atta M. Image-enhanced bronchoscopic evaluation of bronchial mucosal microvasculature in COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2447–55.
Soltani A, Walters E, Reid D, Shukla S, Nowrin K, Ward C, Muller H, Sohal S. Inhaled corticosteroid normalizes some but not all airway vascular remodeling in COPD. Int J Chron Obstruct Pulmon Dis. 2016;11:2359–67.
Harkness L, Kanabar V, Sharma H, Westergren-Thorsson G, Larsson-Callerfelt A. Pulmonary vascular changes in asthma and COPD. Pulm Pharmacol Ther. 2014;29(2):144–55.
Seeger W, Adir Y, Barberà J, Champion H, Coghlan J, Cottin V, De Marco T, Galiè N, Ghio S, Gibbs S, et al. Pulmonary hypertension in chronic lung diseases. J Am Coll Cardiol. 2013;62(25 Suppl):D109–16.
Khanna A, Guo M, Mehra M, Royal W. Inflammation and oxidative stress induced by cigarette smoke in Lewis rat brains. J Neuroimmunol. 2013;254(1–2):69–75.
Lunghi B, De Cunto G, Cavarra E, Fineschi S, Bartalesi B, Lungarella G, Lucattelli M. Smoking p66Shc knocked out mice develop respiratory bronchiolitis with fibrosis but not emphysema. PLoS One. 2015;10(3):e0119797.
Richardson L, Dixon C, Aguilera-Aguirre L, Menon R. Oxidative stress-induced TGF-beta/TAB1-mediated p38MAPK activation in human amnion epithelial cells. Biol Reprod. 2018.
Chen H, Liao K, Cui-Zhao L, Qiang-Wen F, Feng-Zeng X, Ping-Wu F, Liang-Guo S, Juan-Chen Y. Cigarette smoke extract induces apoptosis of rat alveolar type II cells via the PLTP/TGF-β1/Smad2 pathway. Int Immunopharmacol. 2015;28(1):707–14.
Mahmood MQ, Reid D, Ward C, Muller HK, Knight DA, Sohal SS, Walters EH. Transforming growth factor (TGF) beta1 and Smad signalling pathways: a likely key to EMT-associated COPD pathogenesis. Respirology (Carlton, Vic). 2017;22(1):133–40.
Guan S, Liu Q, Han F, Gu W, Song L, Zhang Y, Guo X, Xu W. Ginsenoside Rg1 ameliorates cigarette smoke-induced airway fibrosis by suppressing the TGF-beta1/Smad pathway in vivo and in vitro. Biomed Res Int. 2017;2017:6510198.
Soltani A, Sohal SS, Reid D, Weston S, Wood-Baker R, Walters EH. Vessel-associated transforming growth factor-beta1 (TGF-beta1) is increased in the bronchial reticular basement membrane in COPD and normal smokers. PLoS One. 2012;7(6):e39736.
Ichimaru Y, Krimmer DI, Burgess JK, Black JL, Oliver BG. TGF-beta enhances deposition of perlecan from COPD airway smooth muscle. American journal of physiology Lung cellular and molecular physiology. 2012;302(3):L325–33.
Hogg JC. Pathophysiology of airflow limitation in chronic obstructive pulmonary disease. Lancet. 2004;364(9435):709–21.
Barnes PJ. New concepts in chronic obstructive pulmonary disease. Annu Rev Med. 2003;54:113–29.
Kranenburg AR, de Boer WI, Alagappan VK, Sterk PJ, Sharma HS. Enhanced bronchial expression of vascular endothelial growth factor and receptors (Flk-1 and Flt-1) in patients with chronic obstructive pulmonary disease. Thorax. 2005;60(2):106–13.
Tanimura K, Sato S, Fuseya Y, Hasegawa K, Uemasu K, Sato A, Oguma T, Hirai T, Mishima M, Muro S. Quantitative assessment of erector spinae muscles in patients with chronic obstructive pulmonary disease. Novel chest computed tomography-derived index for prognosis. Annals of the American Thoracic Society. 2016;13(3):334–41.
Pansters NA, Langen RC, Wouters EF, Schols AM. Synergistic stimulation of myogenesis by glucocorticoid and IGF-I signaling. Journal of applied physiology (Bethesda, Md : 1985). 2013;114(9):1329–39.
Lopez IP, Pineiro-Hermida S, Pais RS, Torrens R, Hoeflich A, Pichel JG. Involvement of Igf1r in bronchiolar epithelial regeneration: role during repair kinetics after selective Club cell ablation. PLoS One. 2016;11(11):e0166388.
Ma R, Gong X, Jiang H, Lin C, Chen Y, Xu X, Zhang C, Wang J, Lu W, Zhong N. Reduced nuclear translocation of serum response factor is associated with skeletal muscle atrophy in a cigarette smoke-induced mouse model of COPD. International journal of chronic obstructive pulmonary disease. 2017;12:581–7.
Spruit MA, Gosselink R, Troosters T, Kasran A, Gayan-Ramirez G, Bogaerts P, Bouillon R, Decramer M. Muscle force during an acute exacerbation in hospitalised patients with COPD and its relationship with CXCL8 and IGF-I. Thorax. 2003;58(9):752–6.
Ding H, Wang H, Zhu L, Wei W. Ursolic acid ameliorates early brain injury after experimental traumatic brain injury in mice by activating the Nrf2 pathway. Neurochem Res. 2016.
Rai SN, Yadav SK, Singh D, Singh SP. Ursolic acid attenuates oxidative stress in nigrostriatal tissue and improves neurobehavioral activity in MPTP-induced parkinsonian mouse model. J Chem Neuroanat. 2016;71:41–9.
Kashyap D, Sharma A, Tuli HS, Punia S, Sharma AK. Ursolic acid and Oleanolic acid: Pentacyclic Terpenoids with promising anti-inflammatory activities. Recent Patents Inflamm Allergy Drug Discov. 2016;10(1):21–33.
Wang S, Meng X, Dong Y. Ursolic acid nanoparticles inhibit cervical cancer growth in vitro and in vivo via apoptosis induction. Int J Oncol. 2017;50(4):1330–40.
Yang Y, Li C, Xiang X, Dai Z, Chang J, Zhang M, Cai H, Zhang H, Zhang M, Guo Y, et al. Ursolic acid prevents endoplasmic reticulum stress-mediated apoptosis induced by heat stress in mouse cardiac myocytes. J Mol Cell Cardiol. 2014;67:103–11.
Dong X, Liu S, Zhang L, Yu S, Huo L, Qile M, Liu L, Yang B, Yu J. Downregulation of miR-21 is involved in direct actions of ursolic acid on the heart: implications for cardiac fibrosis and hypertrophy. Cardiovasc Ther. 2015;33(4):161–7.
Lin L, Yin Y, Hou G, Han D, Kang J, Wang Q. Ursolic acid attenuates cigarette smoke-induced emphysema in rats by regulating PERK and Nrf2 pathways. Pulm Pharmacol Ther. 2017;44:111–21.
Murakami S, Takashima H, Sato-Watanabe M, Chonan S, Yamamoto K, Saitoh M, Saito S, Yoshimura H, Sugawara K, Yang J, et al. Ursolic acid, an antagonist for transforming growth factor (TGF)-beta1. FEBS Lett. 2004;566(1–3):55–9.
Yu R, Chen JA, Xu J, Cao J, Wang Y, Thomas SS, Hu Z. Suppression of muscle wasting by the plant-derived compound ursolic acid in a model of chronic kidney disease. J Cachexia Sarcopenia Muscle. 2017;8(2):327–41.
Chen Y, Hanaoka M, Chen P, Droma Y, Voelkel NF, Kubo K. Protective effect of beraprost sodium, a stable prostacyclin analog, in the development of cigarette smoke extract-induced emphysema. American journal of physiology Lung cellular and molecular physiology. 2009;296(4):L648–56.
Li Y, Li J, Li W, Li S, Tian Y, Lu X, Jiang S, Wang Y. Long-term effects of three Tiao-Bu Fei-Shen therapies on NF-κB/TGF-β1/smad2 signaling in rats with chronic obstructive pulmonary disease. BMC Complement Altern Med. 2014;14:140.
Leppäranta O, Myllärniemi M, Salmenkivi K, Kinnula V, Keski-Oja J, Koli K. Reduced phosphorylation of the TGF-Beta signal transducer Smad2 in emphysematous human lung. COPD. 2009;6(4):234–41.
Shin H, Ryu E, Oh E, Kang D. Endoplasmic reticulum stress as a novel target to ameliorate epithelial-to-mesenchymal transition and apoptosis of human peritoneal mesothelial cells. Lab Investig. 2015;95(10):1157–73.
Shah P, Dupre T, Siskind L, Beverly L. Common cytotoxic chemotherapeutics induce epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget. 2017;8(14):22625–39.
Tang X, Liang X, Li M, Guo T, Duan N, Wang Y, Rong G, Yang L, Zhang S, Zhang J. ATF6 pathway of unfolded protein response mediates advanced oxidation protein product-induced hypertrophy and epithelial-to-mesenchymal transition in HK-2 cells. Mol Cell Biochem. 2015;407(1–2):197–207.
Tanjore H, Cheng D, Degryse A, Zoz D, Abdolrasulnia R, Lawson W, Blackwell T. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J Biol Chem. 2015;290(6):3277.
Liang X, Duan N, Wang Y, Shu S, Xiang X, Guo T, Yang L, Zhang S, Tang X, Zhang J. Advanced oxidation protein products induce endothelial-to-mesenchymal transition in human renal glomerular endothelial cells through induction of endoplasmic reticulum stress. J Diabetes Complicat. 2016;30(4):573–9.
Ying R, Wang X, Yang Y, Gu Z, Mai J, Qiu Q, Chen Y, Wang J. Hydrogen sulfide suppresses endoplasmic reticulum stress-induced endothelial-to-mesenchymal transition through Src pathway. Life Sci. 2016;144:208–17.
Acknowledgements
This manuscript has been edited and proofread by Medjaden Bioscience Limited.
Availability of data and material
All data used during the study are available on reasonable request.
Funding
This work was supported by grant from both National Key Technology Research and Development Program of the 13th National 5-year Development Plan (2016YFC1304103) and Liaoning University Innovation Team (LT2013015) program.
Author information
Authors and Affiliations
Contributions
QW participated in the conceived of the study, design of the study and modify of manuscript; JK participated in conceived of the study and modified the manuscript; LL participated in the conceived of the study, carried out the studies, performed the statistical analysis, and draft the manuscript, GH, DH, YY participated in its design, coordination, and perform the statistical analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
We declare that we received approval of the ethics committee of the first affiliated hospital of China medical university for Animal Care and Use in this experiment.
Consent for publication
Not applicable.
Competing interests
The authors declared that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lin, L., Hou, G., Han, D. et al. Ursolic acid alleviates airway-vessel remodeling and muscle consumption in cigarette smoke-induced emphysema rats. BMC Pulm Med 19, 103 (2019). https://doi.org/10.1186/s12890-019-0826-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12890-019-0826-6