
Abstract
Background
Autism entails reduced communicative abilities. Approximately 30% of individuals with autism have intellectual disability (ID). Some people with autism and ID are virtually non-communicative and unable to notify their caregivers when they are in pain. In a pilot study, we showed that heart rate (HR) monitoring may identify painful situations in this patient group, as HR increases in acutely painful situations.
Objectives
This study aims to generate knowledge to reduce the number of painful episodes in non-communicative patients’ everyday lives. We will 1) assess the effectiveness of HR as a tool for identifying potentially painful care procedures, 2) test the effect of HR-informed changes in potentially painful care procedures on biomarkers of pain, and 3) assess how six weeks of communication through HR affects the quality of communication between patient and caregiver.
Methods
We will recruit 38 non-communicative patients with autism and ID residing in care homes. Assessments: HR is measured continuously to identify acutely painful situations. HR variability and pain-related cytokines (MCP-1, IL-1RA, IL-8, TGFβ1, and IL-17) are collected as measures of long-term pain. Caregivers will be asked to what degree they observe pain in their patients and how well they believe they understand their patient’s expressions of emotion and pain. Pre-intervention: HR is measured 8 h/day over 2 weeks to identify potentially painful situations across four settings: physiotherapy, cast use, lifting, and personal hygiene. Intervention: Changes in procedures for identified painful situations are in the form of changes in 1) physiotherapy techniques, 2) preparations for putting on casts, 3) lifting techniques or 4) personal hygiene procedures. Design: Nineteen patients will start intervention in week 3 while 19 patients will continue data collection for another 2 weeks before procedure changes are introduced. This is done to distinguish between specific effects of changes in procedures and non-specific effects, such as caregivers increased attention.
Discussion
This study will advance the field of wearable physiological sensor use in patient care.
Trial registration
Registered prospectively at ClinicalTrials.gov (NCT05738278).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Social communication deficits are among the core features of autism spectrum disorder (ASD) [1]. Symptoms and support requirements for ASD vary in form and intensity with communication deficits ranging from mild to severe [2]. Approximately 30% of people with an ASD diagnosis also have intellectual disability (ID) [3], a neurological condition that affects 1% of the population [4, 5]. For low-functioning individuals with ASD or ID, the symptoms often overlap, and clear-cut diagnosis constitute a challenge [6,7,8]. The diagnosis of ID involves an intelligence quotient (IQ) below 70, reduced adaptive behaviour, and occurrence of the condition before the age of 18 [1, 8]. Approximately 5% of those with ID have grade severe (IQ: 20–34) or profound (IQ < 20) and need round-the-clock supervision and help [9, 10]. Although ID can be an acquired condition, for instance due to cerebral hypoxia, ischemia or infection early in life [11,12,13,14], genetic conditions, such as a de novo mutation [15], are a common cause. Severe or profound ID often coincide with cerebral palsy that affects control of muscles involved in speech, gesticulation, and grimacing. These patients with severe ASD and ID may be unable to notify their caregivers when they are In pain [16,17,18,19,20] and are essentially non-communicative.
There is a need to improve health services for non-verbal patients. Despite the rights of people with disabilities being enshrined in the UN Convention from 2006 [21, 22], age of death remains lower and mortality rates higher for people with intellectual and developmental disabilities [23, 24]. Patients with comorbid ASD and ID often have complex medical needs and many cannot convey their needs to their caregivers [25]. They will typically need the equivalent of 5–10 full-time staff per year–- a major challenge both in terms of competence needs and resource use [26]. Due to the demanding and person-intensive nursing attention required, there is considerable interest in technological solutions to aid communication and participation for this vulnerable group [27, 28]. Overall, technology can improve independence, participation and quality of life among people with complex needs [29].
Sensors may be used to monitor physiological parameters and activity patterns so that individuals and their caregivers can learn about their health. As a neurally mediated phenomenon, heart rate (HR) is regarded a noninvasive window into the central nervous system [30]. Changes in HR are widely used as markers of reactivity to painful events [30,31,32,33,34,35,36,37], and clinically HR is used in neonatal pain assessment and care [31]. Pain is an inferred latent process associated with several physiological and psychological markers including changes in neural activity as measured by electroencephalogram (EEG), pupil dilation, skin conductance, HR, blood pressure, and respiration, as well as self-reported pain magnitude [38, 39]. Although pain-induced increases in blood pressure may trigger vagal baroreflexes that counteract sympathetically-mediated tachycardia; sympathetic changes are found to dominate [40], making HR a reliable non-invasive way to identify probable painful events [40,41,42]. Although the central nervous system, cardiovascular system, and pain are closely interrelated [43, 44], HR is not a specific indicator of pain [30]. States like depression, being tense, angry or frightened may also involve an increase in HR due to a release of stress hormones like cortisol and adrenaline. The clinical application of such reactivity is shown in studies using HR to predict acute stress in ASD [45, 46]. As a more general reactivity measure HR is promising as a tool, and when context is given, it may be useful in non-verbal patient pain-management.
We have previously shown that HR can be used as a person-specific measure of reactivity for non-verbal patients [47]. To evaluate HR as a tool to assist pain management for non-verbal patients, other well-established biomarkers of pain, such as heart rate variability (HRV) and cytokine biomarkers in blood, should be utilized. HRV is a non-invasive measure of imbalances in the autonomic nervous system with a higher variance indicating less stress [48, 49]. A growing body of psychological research has shown HRV to be a stable biomarker of prolonged pain [50], and research supports an association between HRV and emotional responses [51, 52]. Recent studies suggest that certain circulating inflammatory cytokines can be utilized as reliable biomarkers for detecting chronic pain in various diseases [53,54,55]. Blood biomarkers of pain include MCP-1, IL-1RA, IL-8, TGFβ1, and IL-17 [53,54,55]. Elevated levels of these cytokine biomarkers have been associated with experimental pain and self-rated pain, and their levels have been shown to closely follow the temporal phenomenology of pain episodes [54]. The combination of HR, HRV and cytokine biomarkers may prove a novel tool for pain assessment.
Despite the potential of this approach, the use of sensors in care for non-verbal patients is not yet widespread. Most studies on sensors for pain management and communication has been conducted among patients with an ability to communicate [48, 56,57,58,59,60]. Non-verbal patients are frequently left out of research studies, even though advancements in medical technology could bring significant benefits to this patient population.
The proposed study is a randomized controlled trial that seeks to reduce incidence of pain for non-verbal individuals with autism and ID through use of HR-sensors. This study aims to 1) assess the effectiveness of HR as a tool to identify potentially painful care procedures; 2) test the impact of HR-informed changes on pain biomarkers; and 3) examine the impact of six weeks of HR monitoring on the quality of communication between patients and caregivers.
Methods
Study design overview
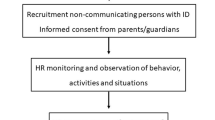
This is a single centre, controlled, randomized trial with 38 non-verbal participants, testing if HR-guided interventions in suspected painful settings will change the incidence of pain. The study design includes a two-week pre-intervention, consisting of continuous HR registering across four potentially painful settings; and an intervention phase consisting of formal change in procedure for the identified painful situation (Fig. 1). The interventions are in one of four forms: changes in 1) physiotherapy techniques, 2) preparations for putting on casts, 3) lifting techniques or 4) personal hygiene procedures. HRV and inflammatory cytokines (MCP-1, IL-1RA, IL-8, TGFβ1, and IL-17) are measured at the beginning, midway, and end of the study period to assess change in long-term pain. Participants will be randomly assigned to either an early intervention group or a delayed intervention group in a 1:1 ratio using a computer-generated randomization scheme. This will differentiate the effect of formal change in routine from increased attentiveness due to study participation, and instant changes made when the HR band signify obvious acute pain (such as skin pinched while putting on a cast). Overview over measures and objectives is given in Table 1.

Overview of phases in a clinical study. Abbreviations: HR: heart rate, HRV: heart rate variability, NCPC: Non-communicating Children’s Pain Checklist; NCAPC: Non-Communicating Adult Pain Checklist
Objectives
The overarching aim is to generate knowledge to reduce incidence of pain in non-verbal patients’ everyday lives.
Primary objective
The primary objective is to evaluate how HR as measure of patient reactivity can be used to identify potentially painful care procedures that should be re-evaluated in terms of the approach taken.
Primary hypothesis: HR increases as defined by 2 standard deviations (SD) increase in HR can identify potentially painful care situations in the patient’s everyday life that that should be re-evaluated (e.g., different methods for stretching of spastic limbs, different routines for transport with personal lifts).
Secondary objectives
The secondary objectives are to:
-
1)
Evaluate how HR-informed changes in potentially painful procedures affect pain biomarkers.
Secondary hypothesis 1: HR-informed changes in patient-specific care procedures (a. physiotherapy, b. applying cast for spastic limbs, c. transportation/being lifted, or d. routines for personal hygiene) will increase HRV and reduce pain-related cytokines (MCP-1, IL-1RA, IL-8, TGFβ1, and IL-17) as evaluated at the beginning, mid-way and at the end of the study period.
-
2)
Assess how six weeks of communication through HR affects the quality of communication between patient and caregiver.
Secondary hypothesis 2: HR as an aid in communication will increase caregivers’ understanding of the patient as measured by changes in perceived understanding of the patient from the beginning to the end of the study period.
Study setting
The study is conducted across several sites in Oslo and the surrounding area (Table 2), with interventions administered at care homes with round-the-clock staff or one-to-one staffed school/daycare. The project management is set in Oslo. We are creating our unique panel of patient representatives, including representatives from LUPE (national user organization for persons with ID, https://www.lupeorg.no/) and the Norwegian Cerebral Palsy Association. The patient representative group will be a national collaboration. The scientific advisory board consist of international researchers working in the field of developmental disability.
Study design
Intervention
The intervention comprises individualized routines to reduce pain in potential pain-inducing settings.
In the pre-intervention phase, HR is continuously collected for 8 h/day across potentially painful care situations. The primary caregiver will report on situations with an HR increase (Figs. 4 and 5).
As the caregiver is not blinded to the HR increases during the registration phase, we expect instantaneous changes made when the HR band communicated obvious acute pain (e.g. skin pinched while putting on cast). These instantaneous, non-formalized changes made by each caregiver is not regarded as “change in routine” but are registered as HR-informed adaptations. The intervention consists of a formal change in routine, and involved changes is written routine and training of staff for the chosen situation. Allocation into an early group and a delayed group helps distinguish the non-formal adaptations based on HR from the effect of formal changes in procedures of care.
Based on the two-week pre-intervention phase, one of four types of care procedures are chosen as the intervention target: 1) physiotherapy, 2) applying cast on spastic limb, 3) transportation/being lifted and 4) personal hygiene procedures. If there are several situations identified as candidates for intervention, selection will be based on a) level of reproducibility and b) magnitude of HR increase. The procedure must be easily recognizable. The situation should at have an HR-spike occurrence for at least 10 episodes during the recording period (i.e. on average 1 occurrence/day), with at least 80% of the situation resulting in HR-increase (reproducibility).
The intervention will be in one of four forms:
-
1)
changes in physiotherapy, e.g., less rigorous movement in the identified painful stretch,
-
2)
preparations for putting on corrective cast to stabilize joint and/or stretch spastic muscles,
-
3)
change in procedures for transportation/lifting, e.g., new technique or adjustments made to equipment, or
-
4)
revised personal hygiene procedure.
If none of the predefined interventions fit the chosen stressor, a fitting intervention is constructed and will be described separately and in detail when the results of this study are published.
Participants
Study Population
Patients with communication difficulties, here defined as an impairment in the ability to convey concepts of both verbal and nonverbal form and that the impairment is to such an extent that it causes caregivers to worry that the patient may experience pain and distress without being able to notify them.
Inclusion criteria
To be included in the study participants must meet the following criteria: Male or female between 5 and 70 years of age at the time of study inclusion. All participants will be evaluated for ASD diagnosis in accordance with ICD-10 by a clinical psychologist taking into consideration that symptoms of ASD and ID often overlap in low-functioning individuals with, making clear-cut diagnosis difficult [6,7,8] (Fig. 2). Social Communication Questionnaire [61,62,63] will be used as part of the diagnostic evaluation. Participants may have cerebral palsy to a degree that compounds the patient’s communication problems. Written informed consent must also be obtained from the participant's legal representative.

Trial flow diagram.As the participant cannot give informed consent, it is the legal representative or parent who decides on participation or to withdraw. Screening includes diagnosis of severe or profound ID and fulfilling ASD diagnostic criteria. Assessments will take place at baseline (week 1), end of week 3 and post-treatment
Exclusion criteria
Not living in a care home with round-the-clock staff for at least five days a week; or not attending one-to-one staffed school/day-care at least five days a week. Having any ongoing infection with a C-reactive protein (CRP) > 20 or any type of cancer with ongoing chemotherapy.
Sample size
There are various approaches for calculating sample size [64]. Here we have decided to design the study to reliably detect our effect size of interest. We will recruit 38 patients, allowing for a dropout of 15%. With 38 patients, a one-sided paired samples t-test can reliably detect (80% power) an effect size of d = 0.84, alpha = 0.05 (Fig. 3). We have chosen a one-sided test as we have a directional hypothesis. This is a realistic effect size of interest given that heart rate increases in response to stressors are associated with effect sizes larger than d = 1 [65].

A power contour plot. Power analysis suggests that a paired samples t-test including 38 participants will reliably detect (power = 80%) an effect size of 0.82, or higher. Statistical power is also shown by assuming smaller or larger hypothetical effect sizes. Analysis and visualisation were performed using the ‘jpower’ JAMOVI module: https://github.com/richarddmorey/jpower
Recruitment
Participants are recruited via Oslo University Hospital, Ahus Hospital, Oslo-, Bærum- and Drammen municipalities, which are located in south-eastern Norway (Table 2). We will invite back people with communication difficulties who have already participated in previous phases of the project two years ago [47], as well as new participants. The investigators will present the study to the local municipalities’ administrators, who will be encouraged to contact the leaders of the districts’ communal residences for persons with ID. Professional caregivers at the care homes will approach the parents or legal representatives of potential participants with written materials describing the study. All recruitment channels will direct the interested individuals to the investigators, and written consent will be obtained from the parents or legally authorized representatives of the participants.
Method of assigning participants to treatment groups
The study design employs an early and a delayed intervention group to distinguish the effects of formal routine changes from increased attentiveness due to study participation and effects of instantaneous changes in care. The early intervention group will start intervention in week three while the delayed group will continue data collection for another two weeks before introducing procedure changes. Patients will be randomly assigned to one of the two groups (Fig. 1) on a 1:1 ratio using a computer-generated randomization scheme.
Blinding
The identity of test and delayed intervention treatments will not be known when analysing data on the secondary outcome measure. Access to the randomization code will be strictly controlled.
Data collection
Clinical assessments
Demographic information (date of birth, gender) will be recorded at inclusion. Relevant medical history, including history of current disease, and information regarding comorbid diagnosis and known aetiology will be recorded at inclusion through conversation with legal representative and primary caregiver, as well as through medical records.
Caregivers’ reports of observed pain
Observable pain is measured using the Non-communicating Children's Pain Checklist (NCPC) for children, and Non-Communicating Adult Pain Checklist (NCAPC) for adults. NCPC is shown to be internally consistent, consistent over time, significantly related to pain intensity, sensitive to pain, and specific to pain [66]. NCAPC has shown equal reliability in assessing chronic pain in adult individuals with ID [67, 68]. Both have proved useful within pain research on participants with ID [67, 69]. Score on NCPC/NCAPC will be recorded at start of trial as this gives each participant a baseline score on how much pain is visible to caregivers prior to use of HR-monitoring.
HR monitoring of acute pain
Polar OH1 or Verity Sense armbands are used to calculate measures of HR [70,71,72,73]. The armband detects pulse at the upper arm (brachial artery), which is transmitted to a Microsoft Surface Pro research laptop (Fig. 4).

Diagram of pipeline. The workflow starts with “HR-band monitoring” registering data, then processing, alerting caregivers, and gathering sensor and situational data for analysis. The final step is the interpretation and eventual publication of results. Abbreviations, HR: heart rate. Icons from flaticon.com
The laptop has a program that calculates whether the HR has risen substantially. This is done by calculating the average HR for the last 20 min. The program then calculates a measure for the variation of the HR within 20 min. If the HR increases by more than 2 standard deviations above the average HR, it sends a signal to alert the caregiver.
Contextual data collection
The research laptop has a registration page for providing contextual data. When an HR increase is detected, the page will automatically appear and ask for information to be provided.
The caregiver will be given four main categories: physiotherapy/stretching, putting on/taking off cast, personal lift use, and hygiene; in addition to “other” with a free text description (Fig. 5). The caregiver checks off stressors according to a pre-made selection of psychological triggers (e.g., excessive stretching, pinched skin, and hot water). Finally, the caregivers check off what signs of pain the patient did show. On all levels there is the possibility of selecting “other” to write in free text.

Context data collection. Presumed triggers for the increased HR is categorized and reported in the computer program during the pre-intervention phase. The caregivers will always have the possibility of reporting freely under “other”. Icons from flaticons.com
HRV monitoring of prolonged pain
HRV is a non-invasive measure of parasympathetic nervous system activity [74, 75]. Lower levels of HRV (i.e., reduced parasympathetic nervous system activity) have been associated with both poor somatic and psychiatric health [76, 77]. Like the HR, HRV is calculated continuously through the Polar OH1 armband. Additionally, HRV will be assessed at rest with a portable electrocardiogram (ECG). The ECG will be collected at baseline, after two weeks of intervention in the early group, and at the end of the study period.
Biochemical measurements
We will collect blood and saliva samples to measure biomarkers for long-term pain and stress at baseline, after two weeks of intervention in the early group, and at the end of the study period.
Determinations for assessment of systemic evidence of inflammation include CRP and ferritin [78], as well as Nam [79, 80]. CRP and Nam will be analysed using marker-specific salivary kits. Blood samples will be analysed at Oslo University Hospital for haemoglobin, haematocrit, red blood cell count, white blood cell count, white blood cell differential, platelet count, erythrocyte sedimentation rate, CRP and ferritin.
Pain-related inflammatory biomarkers include MCP-1, IL-1RA, IL-8, TGFβ1, and IL-17 [81,82,83,84,85,86,87,88]. Analyses will be performed at the Department of Clinical Chemistry, Oslo University Hospital, Oslo, Norway, on an Integra 800 instrument following standard protocol Roche Diagnostics, IN, USA; for details of the diagnostics pipeline, please see Szabo et al., 2022 [89].
PhD A. Szabo will perform all analysis of inflammatory markers at Oslo University Hospital.
Caregivers’ reports of understanding
The participating caregivers will answer an electronic questionnaire on their understanding of the patient and how the caregivers’ level of uncertainty (Table 3). After the study, the caregivers will answer how HR monitoring has affected their understanding of the patient.
The questionnaire is trial specific and based on a review of relevant literature, experience working with the ID population, and discussion within the research group. The questionnaire was tested in a pilot study with 17 participants to perform an exploratory factor analysis for improved psychometric properties. The then-modified questionnaire was distributed to 135 professional caregivers in 2021 [90]. Confirmatory factor analysis indicates good model fit [90].
Strategies to improve adherence to interventions
Measures of treatment compliance
Professional caregivers will be asked to what extent they, due to unplanned events, ended up missing registrations or deviating from the intervention protocol. Before every shift, the caregiver will have to sign a compliance to the intervention protocol, which also operates as a reminder for registrations. The leading caregiver will remind staff of the protocol at every shift meeting. Furthermore, all HR-increases are recorded automatically, and in addition the caregiver will be probed for contextual data for each HR-increase. The discrepancy between number of HR increases and number of contexts recorded gives us a measure of caregiver responsiveness.
Data collection and management
Data management procedures
Participants will not be identified by name in the study database or on any study documents to be collected but will be identified by a site number, participant number, and initials. The principal investigator (Table 2) is responsible for all information collected on participants enrolled in this study. All data collected during this study must be reviewed and verified for completeness and accuracy by the principal investigator.
The data will be entered into Services for sensitive data (TSD): https://www.uio.no/english/services/it/research/sensitive-data/, an enclosed electronic storage system at the University of Oslo. All access to this database are strictly regulated. Only de-identified data will be shared with project team.
All procedures for the handling and analysis of data will be conducted using good computing practices meeting guidelines for the handling and analysis of data for clinical trials.
Data quality control and reporting
All changes to the study database will be documented.
Archival of data
The database is safeguarded against unauthorized access by established security procedures; appropriate backup copies of the database and related software files will be maintained. Databases are backed up by the database administrator in conjunction with any updates or changes to the database.
Statistical methods and considerations
Statistical analyses are performed using SPSS version 25 [92] and R Studio [93].
Demographic and baseline characteristics
The following demographic variables at screening will be summarized: gender and age, height, and weight (for the calculation of body mass index). Descriptive analyses will consist of means and SD for continuous variables and percentages for categorical variables.
Analysis of primary objectives
We hypothesize that a 2 SD increase in HR can identify potentially painful care situations that require re-evaluation. To determine whether HR monitoring is effective in identifying situations that require changes in care, we will compare the mean HR spike counts between situations that are suitable for adjustments in care versus situations that are not. If Shapiro–Wilk test show normality of data distribution, we will use a one-tailed paired sampled t-tests; if non-normality is indicated we will use the Wilcoxon signed-rank test.
The potential for HR to identify potentially painful care procedures will also be shown as descriptions of identified situations and shown as a number count (primary hypothesis). All situations identified by 2 SD increase in HR will be described and reported as count, including an overview of number of times each of the different situations were accompanied by increases in HR (reproducibility); number of HR increases with no registration (responsiveness of caregiver); number of HR increases recorded in situations not previously thought to entail pain or distress (novel discoveries); number of situations discovered through HR-use during the registration phase where instantaneous changes were made due to obvious acute pain (e.g. skin pinched while putting on cast; instantaneous clinical effect of HR); and number of situations identified that were available for adjustments in care and systematic evaluation (formal clinical applicability of HR).
Analysis of secondary objectives
To test evaluate the effect of using HR in improving care procedures and reducing pain, one-tailed paired samples t-tests are used to test for difference in HRV and cytokine biomarkers (MCP-1, IL-1RA, IL-8, TGFβ1, and IL-17) before and after intervention (secondary hypothesis 1). Cohen’s d values [94] will also be calculated to determine the size of the effect. To evaluate the effect of HR-informed changes in procedures, differences in biomarker scores across the early and delayed groups will be analysed using Wilcoxon Signed-rank test, which can evaluate whether the difference scores from each group are not equal (i.e., that they are significantly different). Critical P-values for the five cytokine measurers will be corrected for multiple comparisons using a 5% false discovery rate [95].
To test for significant change in rates of understanding the patient throughout study period (secondary hypothesis 2), we will perform one-tailed paired sampled t-tests. Additionally, Spearman’s rank-order correlation (rs) is used to investigate relationships between HR measures and caregivers report on NCPC and caregivers report on perceived understanding of the patient.
Exploratory post-hoc analysis
With the goal of further developing the technology of the HR-alarm and software, we will do exploratory analysis on how settings of alarm thresholds in the post-processing scripts compare with the event logs.
Administrative, ethical, regulatory considerations
The study will be conducted according to the Declaration of Helsinki.
Minor pain might occur as part of the project; this includes from possible skin reactions to the HR monitor, and pain associated with blood sample collection. For some, the HR monitor can be provocative emotionally, and some might be sensitive to new objects. Ideally, the use of the HR monitor should protect the participant from discomfort.
To maintain confidentiality, all laboratory specimens, evaluation forms, reports and other records will be identified by a coded number and initials only. All study records will be kept in a locked file cabinet and code sheets linking a patient’s name to a patient identification number will be stored separately in another locked file cabinet. Clinical information will not be released without the written permission of the participant. The investigators must also comply with all applicable privacy regulations (e.g., the Health Insurance Portability and Accountability Act of 1996, EU Data Protection Directive 95/46/EC).
Informed consent form
Informed consent will be obtained in accordance with the Declaration of Helsinki. A properly executed, written, informed consent will be obtained from each caregiver to enter the participant into the trial. Information should be given in both oral and written form and legal representatives must be given ample opportunity to inquire about details of the study.
Criteria for discontinuing or modifying allocated interventions
The participants in this study are particularly vulnerable, in that they cannot unequivocally convey how they react to a study measure, such as an HR monitor or bloodwork. It is therefore important to have a high degree of transparency. We believe that we achieve this by involving patient representatives in our project group, and all administrative levels (district director, mayor, agency manager, head of the care homes) in addition to parents, guardians, and caregivers being informed. Several of the mentioned collaborators will, to varying degrees, be involved in the study itself, and the transparency that this entail means security for the participants.
An HR monitor, whether it is attached to the wrist, ankle, or around the chest, can cause skin reactions, both allergic and mechanical. In that case, it must be removed, and an alternative location must be considered, or the use of the HR monitor must be discontinued. Some people with ID and ASD can be very sensitive to new objects. If this applies to the HR monitor, an alternative location must be considered, or the use of the heart rate monitor must be terminated.
As a suitable situation to intervene on is defined by an occurrence of at least 10 for the two-week pre-intervention period, for some participants it might be the case that no suitable situation is identified. In such case, there will be no intervention. However, the patients will be included in the final data presentation, as they shed light on in what proportion of patients HR may be useful for guiding formal change in care.
For caregivers (parents, guardians, family members, and healthcare personnel) the use of HR monitors can potentially distract from other tasks or make the time spent with the non-verbal person feel unnecessarily technical. Alternative placement or termination of the study for the patient in question must be considered against the benefits of using the HR monitor. In cases where it is doubted if caregivers due to such stress do not reliably follow the study protocol, data will be excluded.
A participant may be discontinued from study treatment at any time if the participants’ representatives, the investigators, or the caregivers feel that it is not in the participant’s best interest to continue. Parents or guardians will be able to withdraw the patient from the study without any form of justification.
If a participant is withdrawn from treatment due to an adverse event, the participant will be followed and treated by the principal investigator until the abnormal parameter or symptom has resolved or stabilized.
All participants who discontinue study treatment should come with their parents/guardians for an early discontinuation visit as soon as possible and then should be encouraged to complete all remaining scheduled visits and procedures. Reasonable attempts will be made by the investigators to provide a reason for participant withdrawals. The number and overarching reason for discontinuation or removal of data will be reported in publication of results.
Replacement of participants
Participants who withdraw from the study treatment will not be replaced.
Handling of protocol violation
A protocol violation occurs when the participant or investigators fails to adhere to significant protocol requirements affecting the inclusion, exclusion, participant safety, and primary objectives. Protocol violations for this study include but are not limited to the following: Failure to meet inclusion/exclusion criteria; use of a prohibited concomitant medication; or failure to comply with Good Clinical Practice guidelines will also result in a protocol violation. Number and type of protocol violation will be reported in publication of results.
Discussion
In this randomized blinded controlled trial, we aim to study HR to improve communication between patient and caregiver. This clinical study of HR-informed intervention represents an important contribution to the research and practice of developmental disabilities and communication difficulties. Studies on patients with limited communicative abilities are lacking (Table 4). Existing studies investigating severely limited communicative abilities are small and mainly descriptive. Larger studies on technologies for patients with communication disability exclude the most severely debilitated patients. Furthermore, considerable heterogeneity of participant level of communication disability makes for less clinically relevant findings. In the present study we strive to correct this.
Some limitations of the present study design should be noted. First, HR responses to events are not a specific measure of pain per se [30], and thus must be interpreted in the situational context to be meaningful. However, four potentially painful situations are predetermined, and based on our pilot [47] there is reason to believe a pattern of HR-increase across situations will become evident over a two week period. Furthermore, to bolster the certainty of pain, we complement the HR measures with other extensively studied biomarkers of pain, such as HRV and pain-related cytokines. Second, the caregiver is an intermediary between the researchers and the patient. This means we have no direct objective way to classify situations as all reports of context are given through the caregivers. However, caretakers working with this patient group in Norway are generally well educated and have extensive experience [90], indicating high quality of the observations that are made. We recognize that this indirect method of obtaining contextual information may introduce subjectivity and potential bias. However, we believe that testing the technology through the caregivers is vital to achieving our overarching goal to improve communication and reduce pain in the patient’s everyday life. Setting the study in the patient’s everyday life provides high external validity and ensures that the results are applicable to real-world scenarios. Therefore, while we acknowledge the limitations of our study, we believe that our approach is appropriate and necessary to achieve our research objectives. Third, as the current study targets stressors relevant for each participant, the specific stressors are idiosyncratic. The current design allows for rich individual case data, yet the predefined situations and interventions will provide the benefit of group analysis. This may make the method and design unorthodox and initially more difficult to understand. However, adopting this approach allows for a deeper understanding of the complexity of stressors and interventions as the design permits us to gather exploratory, descriptive, and case-specific data. The current approach provides a unique opportunity to combine the rich individual case data with group trends, leading to a more comprehensive understanding of stressors and interventions.
The ethical implications of this study are complex, particularly regarding the patients’ inability to provide informed consent. The mentioned issue of the caregiver as an intermediary is relevant to the ethical considerations taken when designing the study. An apparent way to bypass the intermediary of the caregiver would be to use video recording in addition to HR. However, HR is generally regarded as less intrusive than video recording and considered more ethically just [97]. If video recordings of caregivers were to be conducted, it would be necessary to obtain informed consent from every caregiver involved, as well as any non-participating patients present in the facility. This would introduce additional ethical considerations related to the proper treatment of human subjects in research. We believe HR is informative yet acceptable, a non-invasive technology, possibly of great beneficence to the patients. Another ethical concern here is the aspect of delayed intervention. Obvious painful situations will be detected during the registration phase, and it is not ethical to postpone making changes in these routines for two or four weeks. This ethical aspect makes for more challenging methodology. However, as these instantaneous adaptations are registered, as well as having a delayed intervention group, results will still be valid for evaluation of HR as a communicative aid.
In conclusion, this trial is a randomized blinded controlled clinical trial that test the applicability of HR to reduce incidence of pain in the participant’s everyday life. We will evaluate how HR can be used to identify potentially painful care procedures that should be re-evaluated in terms of the approach taken; test the effect of HR-informed changes in potentially painful care procedures on biomarkers of pain; and assess how six weeks of communication through HR affects the quality of communication between patient and caregiver. Regardless of outcome, the current study will advance the field of wearable physiological sensor-use in patient care.
Trial status
Inclusion to the study is starting 27th of February 2023. We aim to enrol 38 participants by 2025. The end of data collection will be the end of 2025. Protocol version 1, February 2023.
Availability of data and materials
Not applicable.
Abbreviations
- ID:
-
Intellectual disability
- ASD:
-
Autism spectrum disorder
- HR:
-
Heart rate
- HRV:
-
Heart rate variability
- IQ:
-
Intelligence quotient
- NCPC:
-
Non-communicating Children's Pain Checklist
- NCAPC:
-
Non-Communicating Adult Pain Checklist
- CRP:
-
C-reactive protein
- TSD:
-
Services for sensitive data
- SD:
-
Standard deviations
- ECG:
-
Portable electrocardiogram
- EEG:
-
Electroencephalogram
References
Diagnostic and statistical manual of mental disorders: DSM-5TM, 5th ed. Arlington, VA, US: American Psychiatric Publishing, Inc.; 2013. xliv, 947 p. (Diagnostic and statistical manual of mental disorders: DSM-5TM, 5th ed).
Lord C, Elsabbagh M, Baird G, Veenstra-Vanderweele J. Autism spectrum disorder. Lancet. 2018;392(10146):508–20.
Christensen DL, Baio J, Van Naarden Braun K, Bilder D, Charles J, Constantino JN, et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years--Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. Morb Mortal Wkly Rep Surveill Summ Wash DC 2002. 2016;65(3):1–23.
Zablotsky B, Black LI, Maenner MJ, Schieve LA, Danielson ML, Bitsko RH, et al. Prevalence and trends of developmental disabilities among children in the United States: 2009–2017. Pediatrics. 2019;144(4).
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011;32(2):419–36.
Sappok T, Bergmann T, Kaiser H, Diefenbacher A. Autism in adults with intellectual disabilities. Nervenarzt. 2010;81(11):1333–45.
Heinrich M, Böhm J, Sappok T. Diagnosing autism in adults with intellectual disability: validation of the DiBAS-R in an independent sample. J Autism Dev Disord. 2018;48(2):341–50.
International Classification of Diseases, Eleventh Revision (ICD-11), World Health Organization (WHO) 2019/2021. https://icd.who.int/browse11.
van Bakel M, Einarsson I, Arnaud C, Craig S, Michelsen SI, Pildava S, et al. Monitoring the prevalence of severe intellectual disability in children across Europe: feasibility of a common database. Dev Med Child Neurol. 2014;56(4):361–9.
Smith M, Manduchi B, Burke É, Carroll R, McCallion P, McCarron M. Communication difficulties in adults with Intellectual Disability: Results from a national cross-sectional study. Res Dev Disabil. 2020;97: 103557.
Conway JM, Walsh BH, Boylan GB, Murray DM. Mild hypoxic ischaemic encephalopathy and long term neurodevelopmental outcome - A systematic review. Early Hum Dev. 2018;120:80–7.
Ricci D, Mercuri E, Barnett A, Rathbone R, Cota F, Haataja L, et al. Cognitive outcome at early school age in term-born children with perinatally acquired middle cerebral artery territory infarction. Stroke. 2008;39(2):403–10.
de Montferrand C, Vassel-Hitier J, Yvon-Chaou E, Câmara-Costa H, Dellatolas G, Chevignard M. Language and cognitive outcomes after childhood stroke: Theoretical implications for hemispheric specialization. Cortex J Devoted Study Nerv Syst Behav. 2019;120:509–23.
Ostrander B, Bale JF. Congenital and perinatal infections. Handb Clin Neurol. 2019;162:133–53.
Martin HC, Jones WD, McIntyre R, Sanchez-Andrade G, Sanderson M, Stephenson JD, et al. Quantifying the contribution of recessive coding variation to developmental disorders. Science. 2018;362(6419):1161–4.
Amor-Salamanca A, Menchon JM. Pain underreporting associated with profound intellectual disability in emergency departments. J Intellect Disabil Res JIDR. 2017;61(4):341–7.
Axmon A, Westergren H. Pain and Pain Medication among Older People with Intellectual Disabilities in Comparison with the General Population - PMC. [cited 2022 Oct 23]; Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6023323/
Defrin R, Pick CG, Peretz C, Carmeli E. A quantitative somatosensory testing of pain threshold in individuals with mental retardation. Pain. 2004;108(1–2):58–66.
Boerlage AA, Valkenburg AJ, Scherder EJA, Steenhof G, Effing P, Tibboel D, et al. Prevalence of pain in institutionalized adults with intellectual disabilities: a cross-sectional approach. Res Dev Disabil. 2013;34(8):2399–406.
McGuire BE, Defrin R. Pain perception in people with Down syndrome: a synthesis of clinical and experimental research. Front Behav Neurosci. 2015;9:194.
Konvensjon om rettighetene til personer med nedsatt funksjonsevne [Internet]. [cited 2022 Sep 21]. Available from: https://www.fn.no/om-fn/avtaler/menneskerettigheter/konvensjon-om-rettighetene-til-personer-med-nedsatt-funksjonsevne
Reid SM, Meehan EM, Arnup SJ, Reddihough DS. Intellectual disability in cerebral palsy: a population-based retrospective study. Dev Med Child Neurol. 2018;60(7):687–94.
Lauer E, McCallion P. Mortality of people with intellectual and developmental disabilities from select US state disability service systems and medical claims data. J Appl Res Intellect Disabil JARID. 2015;28(5):394–405.
Doyle A, O’Sullivan M, Craig S, McConkey R. People with intellectual disability in Ireland are still dying young. J Appl Res Intellect Disabil JARID. 2021;34(4):1057–65.
Al-Beltagi M. Autism medical comorbidities. World J Clin Pediatr. 2021;10(3):15–28.
omsorgsdepartementet H og. St.meld. nr. 47 (2008–2009) [Internet]. Regjeringen.no. regjeringen.no; 2009 [cited 2022 Sep 21]. Available from: https://www.regjeringen.no/no/dokumenter/stmeld-nr-47-2008-2009-/id567201/
omsorgsdepartementet H og. Prop. 1 S HOD (2016–2017) [Internet]. Regjeringen.no. regjeringen.no; 2016 [cited 2022 Sep 21]. Available from: https://www.regjeringen.no/no/dokumenter/prop.-1-s-hod-20162017/id2513924/
Ali A, Hassiotis A. Illness in people with intellectual disabilities. BMJ. 2008;336(7644):570–1.
Jamwal R, Jarman HK, Roseingrave E, Douglas J, Winkler D. Smart home and communication technology for people with disability: a scoping review. Disabil Rehabil Assist Technol. 2022;17(6):624–44.
Oberlander T, Saul JP. Methodological considerations for the use of heart rate variability as a measure of pain reactivity in vulnerable infants. Clin Perinatol. 2002;29(3):427–43.
Maxwell LG, Fraga MV, Malavolta CP. Assessment of Pain in the Newborn: An Update. Clin Perinatol. 2019;46(4):693–707.
Hatfield LA, Ely EA. Measurement of acute pain in infants: a review of behavioral and physiological variables. Biol Res Nurs. 2015;17(1):100–11.
Gibbins S, Stevens B. The influence of gestational age on the efficacy and short-term safety of sucrose for procedural pain relief. Adv Neonatal Care Off J Natl Assoc Neonatal Nurses. 2003;3(5):241–9.
Gibbins S, Stevens B, McGrath PJ, Yamada J, Beyene J, Breau L, et al. Comparison of pain responses in infants of different gestational ages. Neonatology. 2008;93(1):10–8.
Mainous RO, Looney S. A pilot study of changes in cerebral blood flow velocity, resistance, and vital signs following a painful stimulus in the premature infant. Adv Neonatal Care Off J Natl Assoc Neonatal Nurses. 2007;7(2):88–104.
Holsti L, Grunau RE. Initial validation of the Behavioral Indicators of Infant Pain (BIIP). Pain. 2007;132(3):264–72.
Holsti L, Grunau RE, Oberlander TF, Osiovich H. Is it painful or not? Discriminant validity of the Behavioral Indicators of Infant Pain (BIIP) scale. Clin J Pain. 2008;24(1):83–8.
Donaldson GW, Chapman RC, Nakamura Y, Bradshaw DH, Jacobson RC, Chapman CN. Pain and the defense response: structural equation modeling reveals a coordinated psychophysiological response to increasing painful stimulation. Pain. 2003;102(1):97.
Kasper D, Fauci A, Hauser S, Longo D, Jameson J, Loscalzo J. Harrison’s principles of Internal Medicine, 19e. Vol. 1. Mcgraw-hill New York:USA; 2015.
Drummond PD. The effect of pain on changes inheart rate during the Valsalva manoeuvre. Clin Auton Res. 2003;13(5):316–20.
Tejman-Yarden S, Levi O, Beizerov A, Parmet Y, Nguyen T, Saunders M, et al. Heart rate analysis by sparse representation for acute pain detection. Med Biol Eng Comput. 2016;54(4):595–606.
Jiang M, Mieronkoski R, Syrjälä E, Anzanpour A, Terävä V, Rahmani AM, et al. Acute pain intensity monitoring with the classification of multiple physiological parameters. J Clin Monit Comput. 2019;33(3):493–507.
Randich A, Maixner W. Interactions between cardiovascular and pain regulatory systems. Neurosci Biobehav Rev. 1984;8(3):343–67.
Foreman RD, Blair RW. Central organization of sympathetic cardiovascular response to pain. Annu Rev Physiol. 1988;50:607–22.
Fioriello F, Maugeri A, D’Alvia L, Pittella E, Piuzzi E, Rizzuto E, et al. A wearable heart rate measurement device for children with autism spectrum disorder. Sci Rep. 2020;10(1):18659.
Nuske HJ, Finkel E, Hedley D, Parma V, Tomczuk L, Pellecchia M, et al. Heart rate increase predicts challenging behavior episodes in preschoolers with autism. Stress Amst Neth. 2019;22(3):303–11.
Kildal E, Stadskleiv K, Boysen ES, Øderud T, Dahl IL, Seeberg TM, et al. Increased heart rate functions as a signal of acute distress in non-communicating persons with intellectual disability. Sci Rep. 2021;19(11):6479.
Acute mental stress assessment via short term HRV analysis in healthy adults: A systematic review with meta-analysis - ScienceDirect [Internet]. [cited 2022 Sep 21]. Available from: https://www.sciencedirect.com/science/article/abs/pii/S1746809415000245
Michels N, Sioen I, Clays E, De Buyzere M, Ahrens W, Huybrechts I, et al. Children’s heart rate variability as stress indicator: Association with reported stress and cortisol. Biol Psychol. 2013;94(2):433–40.
Faye PM, De Jonckheere J, Logier R, Kuissi E, Jeanne M, Rakza T, et al. Newborn infant pain assessment using heart rate variability analysis. Clin J Pain. 2010;26(9):777.
Appelhans BM, Luecken LJ. Heart Rate Variability as an Index of Regulated Emotional Responding. Rev Gen Psychol. 2006;10(3):229–40.
Choi KH, Kim J, Kwon OS, Kim MJ, Ryu YH, Park JE. Is heart rate variability (HRV) an adequate tool for evaluating human emotions? – A focus on the use of the International Affective Picture System (IAPS). Psychiatry Res. 2017;1(251):192–6.
Slade GD, Conrad MS, Diatchenko L, Rashid NU, Zhong S, Smith S, et al. Cytokine biomarkers and chronic pain: association of genes, transcription, and circulating proteins with temporomandibular disorders and widespread palpation tenderness. Pain. 2011;152(12):2802–12.
Jiang X, Zhou R, Zhang Y, Zhu T, Li Q, Zhang W. Interleukin-17 as a potential therapeutic target for chronic pain. Front Immunol [Internet]. 2022 [cited 2023 Jan 12];13. Available from: https://www.frontiersin.org/articles/https://doi.org/10.3389/fimmu.2022.999407
Choi MS, Graves MJ, Matoo S, Storad ZA, Idris RAES, Weck ML, et al. The small EF-hand protein CALML4 functions as a critical myosin light chain within the intermicrovillar adhesion complex. J Biol Chem. 2020;295(28):9281–96.
Koenig J, Jarczok MN, Ellis RJ, Hillecke TK, Thayer JF. Heart rate variability and experimentally induced pain in healthy adults: a systematic review. Eur J Pain Lond Engl. 2014;18(3):301–14.
Picard RW. Future affective technology for autism and emotion communication. Philos Trans R Soc Lond B Biol Sci. 2009;364(1535):3575–84.
Luijcks R, Hermens HJ, Bodar L, Vossen CJ, van Os J, Lousberg R. Experimentally induced stress validated by EMG activity. PLoS One. 2014;9(4).
Kushki A, Drumm E, Pla Mobarak M, Tanel N, Dupuis A, Chau T, et al. Investigating the autonomic nervous system response to anxiety in children with autism spectrum disorders. PLoS One. 2013;8(4).
Empatica | Medical devices, AI and algorithms for remote patient monitoring [Internet]. Empatica. [cited 2022 Sep 21]. Available from: https://www.empatica.com/
Chandler S, Charman T, Baird G, Simonoff E, Loucas T, Meldrum D, et al. Validation of the social communication questionnaire in a population cohort of children with autism spectrum disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(10):1324–32.
Eaves LC, Wingert HD, Ho HH, Mickelson ECR. Screening for autism spectrum disorders with the social communication questionnaire. J Dev Behav Pediatr JDBP. 2006;27(2 Suppl):S95-103.
Berument SK, Rutter M, Lord C, Pickles A, Bailey A. Autism screening questionnaire: diagnostic validity. Br J Psychiatry J Ment Sci. 1999;175:444–51.
Lakens D. Sample Size Justification Collabra Psychol. 2022;8(1):33267.
Reinhardt T, Schmahl C, Wüst S, Bohus M. Salivary cortisol, heart rate, electrodermal activity and subjective stress responses to the Mannheim Multicomponent Stress Test (MMST). Psychiatry Res. 2012;198(1):106–11.
Breau LM, McGrath PJ, Camfield CS, Finley GA. Psychometric properties of the non-communicating children’s pain checklist-revised. Pain. 2002;99(1):349–57.
Weissman-Fogel I, Roth A, Natan-Raav K, Lotan M. Pain experience of adults with intellectual disabilities–caregiver reports. J Intellect Disabil Res JIDR. 2015;59(10):914–24.
Lotan M, Ljunggren EA, Johnsen TB, Defrin R, Pick CG, Strand LI. A modified version of the non-communicating children pain checklist-revised, adapted to adults with intellectual and developmental disabilities: sensitivity to pain and internal consistency. J Pain. 2009;10(4):398–407.
Holmes C, Brock K, Morgan P. Pain and its relationship with postural asymmetry in adults with cerebral palsy: A preliminary exploratory study. Disabil Health J. 2021;14(3): 101063.
Evenson KR, Spade CL. Review of validity and reliability of garmin activity trackers. J Meas Phys Behav. 2020;3(2):170–85.
Schweizer T, Gilgen-Ammann R. Accuracy of the optical heart rate device Polar OH1 during rest and exercise. 2018 [cited 2022 Oct 28]; Available from: https://www.researchgate.net/publication/327261531_Accuracy_of_the_optical_heart_rate_device_Polar_OH1_during_rest_and_exercise
Nunan D, Donovan G, Jakovljevic DG, Hodges LD, Sandercock GRH, Brodie DA. Validity and reliability of short-term heart-rate variability from the Polar S810. Med Sci Sports Exerc. 2009;41(1):243–50.
Lawson LM, Lisk C, Carlson J, Priebe M, Shaver E, Wilson F. The feasibility of measuring heart rate of children with autism during swim lessons and potential health outcomes. Ther Recreation J [Internet]. 2020 Aug 17 [cited 2022 Oct 28];54(3). Available from: https://js.sagamorepub.com/trj/article/view/10200
Akselrod S, Gordon D, Ubel FA, Shannon DC, Berger AC, Cohen RJ. Power spectrum analysis of heart rate fluctuation: a quantitative probe of beat-to-beat cardiovascular control. Science. 1981;213(4504):220–2.
Camm A, Mailk, Bigger. Heart rate variability: standards of measurement, physiological interpretation and clinical use. Task force of the European society of cardiology and the North American society of pacing and electrophysiology. Circulation. 1996;93(5):1043–65.
Kemp AH, Quintana DS. The relationship between mental and physical health: Insights from the study of heart rate variability. Int J Psychophysiol. 2013;89(3):288–96.
Alvares GA, Quintana DS, Hickie IB, Guastella AJ. Autonomic nervous system dysfunction in psychiatric disorders and the impact of psychotropic medications: a systematic review and meta-analysis. J Psychiatry Neurosci. 2016;41(2):89–104.
Bray C, Bell LN, Liang H, Haykal R, Kaiksow F, Mazza JJ, et al. Erythrocyte sedimentation rate and C-reactive protein measurements and their relevance in clinical medicine. WMJ Off Publ State Med Soc Wis. 2016;115(6):317–21.
Martín-Cordero L, García JJ, Hinchado MD, Ortega E. The interleukin-6 and noradrenaline mediated inflammation-stress feedback mechanism is dysregulated in metabolic syndrome: effect of exercise. Cardiovasc Diabetol. 2011;20(10):42.
Pertovaara A. Noradrenergic pain modulation. Prog Neurobiol. 2006;80(2):53–83.
Alvarez P, Green PG, Levine JD. Role for monocyte chemoattractant protein-1 in the induction of chronic muscle pain in the rat. Pain. 2014;155(6):1161–7.
Zhang RX, Li A, Liu B, Wang L, Ren K, Zhang H, et al. IL-1ra alleviates inflammatory hyperalgesia through preventing phosphorylation of NMDA receptor NR-1 subunit in rats. Pain. 2008;135(3):232–9.
Ren K, Torres R. Role of interleukin-1beta during pain and inflammation. Brain Res Rev. 2009;60(1):57–64.
Cunha FQ, Lorenzetti BB, Poole S, Ferreira SH. Interleukin-8 as a mediator of sympathetic pain. Br J Pharmacol. 1991;104(3):765–7.
Sun C, Zhang J, Chen L, Liu T, Xu G, Li C, et al. IL-17 contributed to the neuropathic pain following peripheral nerve injury by promoting astrocyte proliferation and secretion of proinflammatory cytokines. Mol Med Rep. 2017;15(1):89–96.
Vasudeva K, Vodovotz Y, Azhar N, Barclay D, Janjic JM, Pollock JA. In vivo and systems biology studies implicate IL-18 as a central mediator in chronic pain. J Neuroimmunol. 2015;15(283):43–9.
Lantero. Transforming Growth Factor-β in Normal Nociceptive Processing and Pathological Pain Models | SpringerLink. [cited 2022 Oct 18]; Available from: https://link.springer.com/article/https://doi.org/10.1007/s12035-011-8221-1
Echeverry S, Shi XQ, Haw A, Liu g, Zhang Z, Zhang J. Transforming growth factor-β1 impairs neuropathic pain through pleiotropic effects. Mol Pain. 2009;5:1744–8069–5–16.
Szabo A, O’Connell KS, Ueland T, Sheikh MA, Agartz I, Andreou D, et al. Increased circulating IL-18 levels in severe mental disorders indicate systemic inflammasome activation. Brain Behav Immun. 2022;99:299–306.
Kildal E, Hassel B. Service providers’ understanding of the developmentally disabled with large communication difficulties: unfortunate consequences of not understanding each other. In preparation.
Walia S, Wolfe D, Keast D, Ho C, Ethans K, Worley S, et al. Facilitators and barriers for implementing an internet clinic for the treatment of pressure injuries. Telemed J E-Health Off J Am Telemed Assoc. 2019;25(12):1237–43.
IBM SPSS Statistics v. 25.
R: a language and environment for statistical computing.
Feingold A. Effect sizes for growth-modeling analysis for controlled clinical trials in the same metric as for classical analysis. Psychol Methods. 2009;14(1):43–53.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300.
Baxter S, Enderby P, Evans P, Judge S. Interventions using high-technology communication devices: a state of the art review. Folia Phoniatr Logop Off Organ Int Assoc Logop Phoniatr IALP. 2012;64(3):137–44.
Øderud T, Kildal E. Parents’ and professional caregivers’ attitudes towards sensors for identifying pain and distress in non-verbal persons with intellectual disabilit. 2023.
Acknowledgements
Not applicable.
Funding
Open access funding provided by University of Oslo (incl Oslo University Hospital). This research is funded by The Norwegian Research Council, project # 269348 and Stiftelsen Kristian Gerhard Jebsen, project # SKGJ-MED-021.
Author information
Authors and Affiliations
Contributions
All authors (ESMK, DSQ, AS, OA, CT, TN, BH) have contributed substantially to the present work. The first draft was written by ESMK. All authors (ESMK, DSQ, AS, OA, CT, TN, BH) have added to the previous versions of the manuscript. All authors (ESMK, DSQ, AS, OA, CT, TN, BH) have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Regional Committees for Medical and Health Research Ethics South East Norway (approval #2016/1956, #2009/932 and #2012/1967). All research will be carried out in accordance with the Declaration of Helsinki. Written informed consent will be obtained from the participant's legal representative.
Consent for publication
Not applicable.
Competing interests
No conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kildal, E.S.M., Quintana, D.S., Szabo, A. et al. Heart rate monitoring to detect acute pain in non-verbal patients: a study protocol for a randomized controlled clinical trial. BMC Psychiatry 23, 252 (2023). https://doi.org/10.1186/s12888-023-04757-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12888-023-04757-1