
Abstract
Background
Most patients with Posttraumatic Stress Disorder (PTSD) suffer residual symptoms following first-line treatment. Oxidative stress has been implicated in the pathophysiology of PTSD. N-acetylcysteine (NAC) is a precursor of the brain’s primary antioxidant, glutathione, and may diminish oxidative cellular damage. An 8-week pilot study of NAC in veterans with PTSD found that symptoms were significantly reduced in the NAC group compared to placebo. This study aims to confirm these findings with a larger sample in a double-blind, placebo-controlled trial to further explore the efficacy of NAC as an adjunctive therapy in treatment-resistant PTSD.
Methods
A multicentre, randomised, double-blind, placebo-controlled trial for adult patients who still meet criteria for PTSD following first-line treatment. The intervention comprises either NAC as a fixed dose regime of 2.7 g/day (900 mg three times daily) administered orally for 12 weeks, or placebo. Standard care for PTSD will continue in addition, including other pharmacotherapies. Detailed clinical data will be collected at randomisation and weeks 4, 8, 12, 16, and 64 post-randomisation, with self-report measures completed weekly from baseline to 16 weeks and at 64 weeks post-randomisation. Blood-based biomarkers will be collected at baseline and 12 weeks to assess the mechanism of effect. The primary outcome measure will be change in Clinician-Administered PTSD Scale for DSM-5 at 12 weeks compared with baseline. Secondary outcomes will be change in quality of life, depression, anxiety, substance use and craving, and somatic symptoms. With 126 completed participants (63 per arm), the study is powered at 80% to detect a true difference in the primary outcome measure using a two-tailed analysis with alpha = 0.05, beta = 0.2.
Discussion
This is the first multicentre, double blind, randomised, placebo-controlled trial of adjunctive NAC for treatment-resistant PTSD. NAC has an established safety profile, is readily available and easy to administer, and has a favourable tolerability profile, therefore making it an attractive adjunctive therapy. Inclusion of blood analyses to assess potential target engagement biomarkers of oxidative stress and neuroinflammation may help gauge the biological mechanisms of effect of NAC.
Trial registration
ACTRN12618001784202, retrospectively registered 31/10/2018, URL: http://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=376004.
Similar content being viewed by others


Background
Posttraumatic Stress Disorder (PTSD) is a condition characterized by re-experiencing of trauma, deterioration in cognition and mood, avoidance of triggers and hyper-arousal symptoms after experiencing severely threatening, traumatic events [1]. Large community surveys suggest that most people will experience at least one potentially traumatic event in their lives [2], however in a proportion of individuals, symptoms of psychological distress persist and progress to the development of PTSD. PTSD affects approximately 6% of the population, though rates are considerably higher for certain occupational groups such as emergency service workers and military personnel [3]. It is difficult to treat, with only a quarter of patients achieving full remission with first-line therapy [4].
PTSD is associated with substantial disability as symptoms are usually chronic [5] and accompanied by extensive functional decline, including high rates of unemployment [6], family and relationship difficulties [7], suicidality [8] and reduced quality of life [9]. PTSD is also frequently associated with a variety of comorbidities such as depression, substance abuse and psychosomatic disorders [1]. Recommended first-line treatments of PTSD in Australia are psychological, including trauma-focused psychotherapy, but they suffer from limited tolerability, and many patients discontinue treatment prematurely [10]. Selective-serotonin reuptake inhibitors (SSRIs) are the preferred pharmacological intervention for PTSD, either as alone or in conjunction with psychological therapies [11, 12]. SSRIs have demonstrated only moderate efficacy, however, with the majority of patients failing to achieve remission even when combined with psychotherapy [13, 14]. Those who do not respond to first-line treatment are usually considered treatment-resistant, and by this measure up to 70% of patients with PTSD fall into that category [15].
Several lines of evidence suggest that increased oxidative stress and neuroinflammation are implicated in the pathophysiology of PTSD [16,17,18]. Oxidative stress occurs when there are excess levels of free radicals, or inadequate antioxidants, potentially leading to cellular damage. Multiple studies in PTSD samples have demonstrated elevated levels of biomarkers of oxidative-stress, including anti-oxidant blood-based enzyme concentrations and altered gene expression [19, 20]. The brain’s primary antioxidant, glutathione, is dysregulated in patients with PTSD, [21] and downregulation of the Glutathione-S-Transferase Mu1 & 2 genes, responsible for encoding enzymes involved in conjugation reactions with glutathione, is considered the most reliable blood-based biomarker for PTSD [22]. Replenishing glutathione could be expected to diminish oxidative cellular damage, and hence potentially ameliorate PTSD symptoms and prevent long-term illness sequelae. However, glutathione is not orally bioavailable. In order to restore glutathione supply, its synthesis must be enhanced, for example by a precursor such as N-acetylcysteine (NAC).
Research indicates that NAC, or more likely its metabolites, can cross the blood-brain barrier [23, 24] and systemic delivery leads to a rise in brain glutathione [25]. While emerging evidence supports the use of NAC in the treatment of other psychiatric disorders [26, 27], this has yet to be thoroughly explored in PTSD. A small pilot study assessing NAC as an adjunct to cognitive-behavioural therapy in a group of US veterans with co-morbid substance use disorders demonstrated significant reductions in self-reported PTSD symptoms, co-morbid depressive symptoms and cravings for substance use compared to the placebo group. In view of these findings and the growing understanding of theoretical foundations of oxidation biology, NAC appears well placed as a potential adjunctive treatment for PTSD. It is well-tolerated [28, 29], low cost and is available without prescription. To date, NAC has yet to be evaluated in a large scale, randomised clinical trial as an adjuvant treatment for PTSD.
Study objectives
This study aims to investigate the efficacy of adjunctive NAC for patients with treatment-resistant PTSD. It is hypothesised that NAC plus treatment as usual, will be superior to placebo plus treatment as usual in reducing clinician-assessed PTSD symptoms in an adult PTSD sample. It is also hypothesised that adjunctive NAC will result in a reduction in self-reported PTSD, depressive and somatic symptoms, substance use and cravings, and improvement in quality of life. This study will also explore blood-based biomarkers in PTSD to elucidate the mechanisms of action of NAC on PTSD symptoms.
Methods
Study design and setting
This study is a multi-site, 12-week, double-blind, randomised, placebo-controlled trial. The trial will be conducted at two tertiary mental-health centres in Australia which provide specialist PTSD treatment services: Austin Health’s public Psychological Trauma Recovery Service (PTRS) and The Melbourne Clinic (TMC) which is a private psychiatric hospital. Both sites acquired ethics approval to allow recruitment of participants from their clinical services and from the general community.
Patient and public involvement
Patients or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this study.
Patients
Eligibility criteria are outlined in Table 1. The target population includes individuals aged 18 and older who meet Diagnostic & Statistical Manual of Mental Disorders - Fifth edition (DSM-5) diagnostic criteria for current PTSD despite having undergone a recommended course of treatment. A treatment course is defined as a full course of trauma-focused psychotherapy or a 6-week (minimum) course of pharmacotherapy (SSRI or SNRI). Participants may continue their usual treatments during the study. Participants must have the capacity to consent to the study and to follow its instructions and procedures.
Sample size calculation and recruitment target
The target sample size is 126 participants (63 per arm). For a two-tailed analysis with alpha = 0.05, beta = 0.2, the study is powered at 80% to detect a true difference in CAPS-5 scores between the NAC and placebo groups of 10+/− 20 points. In our pilot trial, effect sizes for PTSD symptoms as well as all secondary measures exceeded this threshold. Using a conservative attrition rate of one-third, we will need to recruit 190 participants to have 126 complete the trial and follow-up.
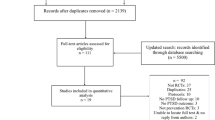
The study aims to recruit participants referred by their treating clinician or self-referred from the community. A summary of the participant flow through the study is shown in Fig. 1.

Trial Flow Diagram. NAC – N-Acetyl Cysteine; RT – Routine treatment; PCL-5 – PTSD Checklist 5
Screening phase and baseline assessment
Individuals with PTSD who express interest in participating in the study will be contacted by a trial clinician and screened according to the eligibility criteria outlined in Table 1 via a brief telephone interview.
After initial telephone screening, eligible participants will be invited to a face-to-face or videoconference 90-min clinical interview. After obtaining IRB-approved written informed consent, a diagnosis of PTSD will be confirmed through the Clinician-Administered PTSD Scale for DSM-5 (CAPS-5) – monthly version, which is the gold-standard in PTSD assessment. Participants will be excluded if they do not meet diagnostic criteria for current PTSD. If patients are deemed eligible and are enrolled in the study, this interview will constitute part of the baseline assessment.
The baseline assessment will also include data collection via clinician-administered and self-reported questionnaires regarding psychiatric comorbidities, PTSD symptoms, quality of life, substance use and somatic symptoms (see Table 2). Detailed clinical and demographic information will be collected on all participants at the baseline clinical interview. This will include age, gender, marital status, years of formal education, service history, receipt of state benefits, ethnicity, and country of birth.
Participants will be provided with a signed copy of their information and consent form which includes written information regarding study protocol, purpose, data handling, participant confidentiality, potential side-effects and their right to withdraw from the study at any time.
Allocation
Randomisation will take place only once participants have undertaken the baseline assessment and are confirmed eligible for the trial. If all inclusion and exclusion criteria are satisfied, trial participants will be randomised at a 1:1 ratio to the NAC (treatment) or placebo arm. Random block allocation of medication packs will be in a four-to-a-block design, randomly generated by a computer program. Randomisation will be carried out by the clinical trial pharmacy service at the trial site. Allocation will be concealed from participants and all other research staff as the study is double blinded.
A fixed dose regime of 2.7 g/day of NAC will be administered as 900 mg three times daily for 12 weeks. Previous pilot studies have demonstrated this dose is efficacious and well-tolerated [28, 29]. The NAC and placebo are either supplied as effervescent tablets or as capsules. The trial medications will be supplied on a monthly basis (to coincide with clinical interviews) and participants will be instructed to return all packaging to allow capsule counts by the trial monitor and the trial pharmacist. Adherence will be assessed by pill counts of returned medication packs. Participants will also be required to complete a daily pill diary to document treatment compliance. The trial medications will be supplied, and returned containers counted, by the trial pharmacist.
Blinding
To facilitate the double-blinding process, the trial medications (both NAC and placebo) will be dispensed in identical numbers and tablet forms in sealed containers, and the placebo containers will be specially treated with microgram levels of NAC dust to produce its characteristic smell. Placebo will be manufactured according to European Medicine Agency (EMA) guidelines. We will require research workers to indicate if they have become unblinded so that outcome assessments can be undertaken by blinded assessors in addition to any established treatments for their PTSD.
Follow-up assessments
Following baseline assessment and enrolment in the trial, participants will be assessed with clinical interviews at weeks 4, 8 and 12 of the trial. Post-trial interviews are conducted at 16 weeks and 64 weeks to assess retention of any treatment effects. At each follow-up assessment, clinician-rated PTSD symptoms, and self-reported PTSD symptoms as well as psychiatric comorbidities, quality of life, substance use and somatic symptoms are assessed (see Table 2). In between follow-up interviews (from Baseline to Week 16), participants will complete weekly self-report questionnaires regarding their PTSD symptoms. Follow-up interviews will take about 60 min to complete; the weekly self-report questionnaire take approximately 5 min.
Outcomes
Our primary evaluation of treatment efficacy is the reduction of observer-rated PTSD symptom severity, operationalised as a reduction CAPS-5 severity scores. Secondary objectives include evaluation of NAC to reduce self-reported symptoms of (1) PTSD, (2) depression and anxiety, (3) substance use, (4) craving and (5) somatic symptoms. Evaluation of treatment efficacy will be assessed over the entire trial period. Retention of treatment effects will be assessed at weeks 16 and 64 post-randomisation. The safety and tolerability of the intervention will be evaluated by self-report.
Participants may elect to provide a blood sample at baseline and Week 12 assessments. Blood based biomarkers of oxidative stress will be evaluated as exploratory outcomes. Blood analysis will include measurement of IL-6 (interleukin-6), TNF-alpha (tumour necrosis factor alpha), TBARS (thiobarbituric acid reactive substances), MDA (malondialdehyde) and BDNF (brain-derived neurotrophic factor). Modulation of these blood-based proteins will be correlated with treatment response. Participants will not be fasting prior to the blood collection. BD vacutainer SST II advance tubes (5 mL) will be used to collect the sample and then allowed to rest for 30 min prior to being centrifuged at 1300 g for 15 mins. The supernatant (serum) will be extracted and aliquoted into 2 × 1.8 ml tubes. All samples will be stored at − 80 degrees Celsius until tested.
Participant safety and withdrawal
Adverse effects will be assessed each week and reviewed by the study team as well as the Data Safety Monitoring Board, comprising the investigators. Though serious adverse events with NAC are extremely rare, patients will be advised to seek appropriate emergency medical help in such an instance. For less serious events, participants will be advised to contact their treating physician as usual. Participants will be withdrawn from the trial under the following conditions: (i) failure to take the trial medication for seven consecutive days; (ii) cessation of effective contraception or pregnancy; and (iii) withdrawal of consent or emergence of adverse events (as determined by the medical investigators or decision of the participant). All data will be retained in the intention-to-treat analysis.
Standardised medical care
Standard care for participants will involve their regular, medical appointments at variable intervals. The additional procedures involve meeting with the research clinician in outpatients of the clinical services – where possible to coincide with scheduled appointments – at 4-weekly intervals, where they will engage in a clinical interview, and complete self-report questionnaire items. Participants will also be asked to complete one self-report questionnaire, each week on their own.
Interrater reliability
All research staff undertaking clinical assessments will be trained in the assessments prior to commending recruitment. All clinical research staff will undertake observed interviews on a yearly basis to ensure compliance with standardised administration guidelines and complete reliability and inter-rater reliability assessments.
Statistical analysis
All analyses will be conducted by our independent trial statistician and will include all randomised participants with at least one post-baseline observation (modified intention to treat). The statistician will be blind to group allocation (triple blind design). Differences between study arms at baseline will be assessed using either chi-squared or Fisher’s exact test for categorical data and either student t–test or Mann-Whitney U test for continuous data. Non-parametric statistics will be used when assumptions for parametric methods are violated. Effect sizes will be calculated using Cohen’s guidelines. The primary efficacy analysis will assess the impact of the treatment on group differences for the primary outcome measure (CAPS-5) over the entire study period and use a likelihood based mixed-effects model, repeated measures approach (MMRM). The MMRM model includes the categorical effects of treatment, investigator, visit, and treatment-by-visit interaction, as well as the continuous, fixed covariates of baseline score and baseline score-by-visit interaction.
Data handling and monitoring
Paper copies of the questionnaires will be kept in a locked filing cabinet in the Department of Psychiatry of the clinical services, in a room kept locked. The data will be entered into a computer in re-identifiable form, with the code kept in a separate file. Computer files will be kept on a password-protected computer in the department of psychiatry in a room kept locked at night. All files will be kept for 15 years before secure disposal.
All blood samples will be securely stored at the Austin Health Pathology Department in a monitored, trial specific freezer at − 80 degrees Celsius and labelled in a re-identifiable format before being transferred to the Flory Institute of Neuroscience and Mental Health for analysis. Following analysis, all blood will be securely destroyed by the Flory Institute of Neuroscience and Mental Health. Blood samples collected for the purposes of this study that are not transferred to the Flory Institute of Neuroscience and Mental Health for analysis will be destroyed after a period of 7 years.
Discussion
This study described the design and methodology of the first multi-centre, randomised, double-blind, placebo-controlled 12-week trial evaluating the efficacy of NAC as an adjuvant therapy in treatment-resistant PTSD. Despite recent advancements in evidence-based interventions for PTSD, many patients continue to suffer substantial residual symptoms post-treatment [13, 14, 38]. Accumulating evidence supports the potential efficacy of NAC in symptom reduction and functional improvements across a range of psychiatric and neurological disorders [26, 27, 39]. We hypothesise that when delivered as an adjuvant therapy, NAC will reduce PTSD and related symptomatology greater than treatment as usual.
Oral NAC is widely available over the counter as an inexpensive nutritional supplement and has demonstrated low rates of adverse effects [26, 39, 40]. These factors support the use of NAC in clinical settings as a safe, inexpensive, easily administered and non-invasive supplementary treatment strategy for PTSD. However, recommendations regarding the use of NAC in psychiatric disorders are limited by the small number, small sample sizes and quality of existing studies [26]. As such, large controlled trials assessing the efficacy of NAC in specific disorders are essential to establish its efficacy. The anticipated sample size of the present study is likely to exceed many previous clinical studies [41], thus increasing the statistical power and generalizability of the study findings. The large sample size, and randomised placebo-controlled design are significant methodological strengths in the current study. A further strength is the assessment of clinical outcomes at 4- and 52-weeks post-trial completion. Previous clinical trials have produced mixed results regarding the retention of treatment benefits of NAC in other psychiatric disorders, with some effects either retaining or emerging at 4 weeks post-trial completion and others being lost [29, 42].
While NAC has been implicated as an effective adjunctive treatment for many psychiatric conditions, the mechanism of effect has not yet been fully elucidated. The therapeutic effects of NAC are likely mediated via its ability to modulate numerous pathophysiological processes including oxidative stress, neurogenesis and apoptosis, mitochondrial dysfunction, neuroinflammation and dysregulation of glutamate and dopamine [26]. Determining the underlying mechanisms of NAC in PTSD is helpful, not only to understanding the underlying neurobiology of the illness but to discover additional therapies that may operate on similar pathophysiological pathways. Of key interest in the current study is NAC’s ability to replenish glutathione and thereby diminish oxidative stress and neuroinflammation, which are well established biomechanisms in PTSD and its other psychiatric comorbidities [16,17,18]. While studies have speculated on the mechanisms of action of NAC on psychiatric symptoms [26, 40] few have directly assessed blood-based biomarkers of oxidative stress and neuroinflammation in the context of treatment with NAC. The inclusion of blood-based biomarkers of oxidative stress and neuroinflammation is therefore a key methodological advantage of the present study.
A final advantage for this study is the heterogeneity of the sample. The broad inclusion and limited exclusion criteria mean that participants experiencing a range of psychiatric comorbidities will be eligible, and the adjuvant approach means that it will be available to most individuals with PTSD. To overcome the lack of specificity incurred by the more ecologically valid inclusion criteria, this study will thoroughly characterise psychiatric comorbidities and medical histories at baseline, so as to be able to distinguish whether treatment responses vary between presentations. Use of the gold standard CAPS-5 PTSD diagnostic tool will also enable the study to characterise trauma histories, and establish whether treatment responses differ between trauma types. This is particularly important, given that PTSD symptom severity and treatment response rates may differ between those with a history of prolonged developmental trauma and those with combat related PTSD [43,44,45,46].
Availability of data and materials
Anonymised, participant level data will be available from the corresponding author on reasonable request.
Abbreviations
- PTSD:
-
Posttraumatic Stress Disorder
- PTRS:
-
Posttraumatic Recovery Service
- TMC:
-
The Melbourne Clinic
- NAC:
-
N-acetylcysteine
- CAPS-5:
-
Clinical Administered PTSD scale for DSM-5
References
Association AP. Diagnostic and statistical manual of mental disorders. 5th ed; 2013.
Mills KL, McFarlane AC, Slade T, Creamer M, Silove D, Teesson M, et al. Assessing the prevalence of trauma exposure in epidemiological surveys. Aust N Z J Psychiatry. 2011;45(5):407–15.
Milliken CS, Auchterlonie JL, Hoge CW. Longitudinal assessment of mental health problems among active and reserve component soldiers returning from the Iraq war. JAMA. 2007;298(18):2141–8.
Berger W, Mendlowicz MV, Marques-Portella C, Kinrys G, Fontenelle LF, Marmar CR, et al. Pharmacologic alternatives to antidepressants in posttraumatic stress disorder: a systematic review. Prog Neuro-Psychopharmacol Biol Psychiatry. 2009;33(2):169–80.
Department of Veterans Affairs and the Department of Defense. VA/DoD Clinical practice guideline for the management of posttraumatic stress disorder and acute stress disorder, 3.0; 2017. p. 13–14.
Savoca E, Rosenheck R. The civilian labor market experiences of Vietnam-era veterans: the influence of psychiatric disorders. J Ment Health Policy Econ. 2000;3(4):199–207.
Riggs DS, Byrne CA, Weathers FW, Litz BT. The quality of the intimate relationships of male Vietnam veterans: problems associated with posttraumatic stress disorder. J Trauma Stress. 1998;11(1):87–101.
Krysinska K, Lester D. Post-traumatic stress disorder and suicide risk: a systematic review. Arch Suicide Res. 2010;14(1):1–23.
Buckley TC, Mozley SL, Bedard MA, Dewulf AC, Greif J. Preventive health behaviors, health-risk behaviors, physical morbidity, and health-related role functioning impairment in veterans with post-traumatic stress disorder. Mil Med. 2004;169(7):536–40.
Gros DF, Tuerk PW, Yoder M, Acierno R. Handbook of clinical psychology competencies, volume II: intervention and treatment for adults. New York: Springer; 2011.
Cooper J, Metcalf O, Phelps A. PTSD--an update for general practitioners. Aust Fam Physician. 2014;43(11):754–7.
Health PACfPM. Australian guidelines for the treatment of acute stress disorder and posttraumatic stress disorder. Melbourne: Phoenix Australia; 2013.
Hamner MB, Robert S, Frueh BC. Treatment-resistant posttraumatic stress disorder: strategies for intervention. CNS Spectr. 2004;9(10):740–52.
Davidson JR. Pharmacologic treatment of acute and chronic stress following trauma: 2006. J Clin Psychiatry. 2006;67(Suppl 2):34–9.
Starke JA, Stein DJ. Management of Treatment-Resistant Posttraumatic Stress Disorder. Curr Treat Options Psych. 2017;4:387–403.
Bremner JD. Stress and brain atrophy. CNS Neurol Disord Drug Targets. 2006;5(5):503–12.
Baker DG, Nievergelt CM, O'Connor DT. Biomarkers of PTSD: neuropeptides and immune signaling. Neuropharmacology. 2012;62(2):663–73.
Furtado M, Katzman MA. Neuroinflammatory pathways in anxiety, posttraumatic stress, and obsessive compulsive disorders. Psychiatry Res. 2015;229(1–2):37–48.
Atli A, Bulut M, Bez Y, Kaplan I, Ozdemir PG, Uysal C, et al. Altered lipid peroxidation markers are related to post-traumatic stress disorder (PTSD) and not trauma itself in earthquake survivors. Eur Arch Psychiatry Clin Neurosci. 2016;266(4):329–36.
Borovac Stefanovic L, Kalinic D, Mimica N, Beer Ljubic B, Aladrovic J, Mandelsamen Perica M, et al. Oxidative status and the severity of clinical symptoms in patients with post-traumatic stress disorder. Ann Clin Biochem. 2015;52(Pt 1):95–104.
Michels L, Schulte-Vels T, Schick M, O'Gorman RL, Zeffiro T, Hasler G, et al. Prefrontal GABA and glutathione imbalance in posttraumatic stress disorder: preliminary findings. Psychiatry Res. 2014;224(3):288–95.
Tylee DS, Chandler SD, Nievergelt CM, Liu X, Pazol J, Woelk CH, et al. Blood-based gene-expression biomarkers of post-traumatic stress disorder among deployed marines: a pilot study. Psychoneuroendocrinology. 2015;51:472–94.
Farr SA, Poon HF, Dogrukol-Ak D, Drake J, Banks WA, Eyerman E, et al. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84(5):1173–83.
Valdovinos-Flores C, Gonsebatt ME. The role of amino acid transporters in GSH synthesis in the blood-brain barrier and central nervous system. Neurochem Int. 2012;61(3):405–14.
Dean OM, van den Buuse M, Berk M, Copolov DL, Mavros C, Bush AI. N-acetyl cysteine restores brain glutathione loss in combined 2-cyclohexene-1-one and d-amphetamine-treated rats: relevance to schizophrenia and bipolar disorder. Neurosci Lett. 2011;499(3):149–53.
Deepmala, Slattery J, Kumar N, Delhey L, Berk M, Dean O, et al. Clinical trials of N-acetylcysteine in psychiatry and neurology: A systematic review. Neurosci Biobehav Rev. 2015;55:294–321.
Ooi SL, Green R, Pak SC. N-Acetylcysteine for the treatment of psychiatric disorders: a review of current evidence. Biomed Res Int. 2018;2018:2469486.
Berk M, Copolov D, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N-acetyl cysteine as a glutathione precursor for schizophrenia--a double-blind, randomized, placebo-controlled trial. Biol Psychiatry. 2008;64(5):361–8.
Berk M, Copolov DL, Dean O, Lu K, Jeavons S, Schapkaitz I, et al. N-acetyl cysteine for depressive symptoms in bipolar disorder--a double-blind randomized placebo-controlled trial. Biol Psychiatry. 2008;64(6):468–75.
Weathers FW, Bovin MJ, Lee DJ, Sloan DM, Schnurr PP, Kaloupek DG, et al. The clinician-administered PTSD scale for DSM-5 (CAPS-5): development and initial psychometric evaluation in military veterans. Psychol Assess. 2018;30(3):383–95.
Zigmond AS, Snaith RP. The hospital anxiety and depression scale. Acta Psychiatr Scand. 1983;67(6):361–70.
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62.
Blevins CA, Weathers FW, Davis MT, Witte TK, Domino JL. The posttraumatic stress disorder checklist for DSM-5 (PCL-5): development and initial psychometric evaluation. J Trauma Stress. 2015;28(6):489–98.
Kroenke K, Spitzer RL, Williams JB. The PHQ-15: validity of a new measure for evaluating the severity of somatic symptoms. Psychosom Med. 2002;64(2):258–66.
Skevington SM, Lotfy M, O'Connell KA, Group W. The World Health Organization's WHOQOL-BREF quality of life assessment: psychometric properties and results of the international field trial. A report from the WHOQOL group. Qual Life Res. 2004;13(2):299–310.
Saunders JB, Aasland OG, Babor TF, de la Fuente JR, Grant M. Development of the alcohol use disorders identification test (AUDIT): WHO collaborative project on early detection of persons with harmful alcohol consumption--II. Addiction. 1993;88(6):791–804.
Sheehan DV, Lecrubier Y, Sheehan KH, Amorim P, Janavs J, Weiller E, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998;59(Suppl 20):22–33 quiz 4–57.
Steenkamp MM, Litz BT, Hoge CW, Marmar CR. Psychotherapy for military-related PTSD: a review of randomized clinical trials. JAMA. 2015;314(5):489–500.
Fernandes BS, Dean OM, Dodd S, Malhi GS, Berk M. N-Acetylcysteine in depressive symptoms and functionality: a systematic review and meta-analysis. J Clin Psychiatry. 2016;77(4):e457–66.
Berk M, Malhi GS, Gray LJ, Dean OM. The promise of N-acetylcysteine in neuropsychiatry. Trends Pharmacol Sci. 2013;34(3):167–77.
Back SE, McCauley JL, Korte KJ, Gros DF, Leavitt V, Gray KM, et al. A double-blind, randomized, controlled pilot trial of N-Acetylcysteine in veterans with posttraumatic stress disorder and substance use disorders. J Clin Psychiatry. 2016;77(11):e1439–e46.
Berk M, Dean OM, Cotton SM, Jeavons S, Tanious M, Kohlmann K, et al. The efficacy of adjunctive N-acetylcysteine in major depressive disorder: a double-blind, randomized, placebo-controlled trial. J Clin Psychiatry. 2014;75(6):628–36.
Bradley R, Greene J, Russ E, Dutra L, Westen D. A multidimensional meta-analysis of psychotherapy for PTSD. Am J Psychiatry. 2005;162(2):214–27.
De Jongh A, Resick PA, Zoellner LA, van Minnen A, Lee CW, Monson CM, et al. Critical analysis of the current treatment guidelines for complex Ptsd in adults. Depress Anxiety. 2016;33(5):359–69.
Watts BV, Schnurr PP, Mayo L, Young-Xu Y, Weeks WB, Friedman MJ. Meta-analysis of the efficacy of treatments for posttraumatic stress disorder. J Clin Psychiatry. 2013;74(6):e541–50.
Wolf EJ, Miller MW, Kilpatrick D, Resnick HS, Badour CL, Marx BP, et al. ICD-11 complex PTSD in US national and veteran samples: prevalence and structural associations with PTSD. Clin Psychol Sci. 2015;3(2):215–29.
Acknowledgements
Not applicable.
Funding
This work was supported by the Defence Health Foundation, who peer-reviewed the study, and the Sir Edward Dunlop Medical Research Foundation. The NAC and placebo for the first 30 participants were donated by Bioadvantex. Neither the funders nor the sponsor had any role in the design, execution, analysis or publication of this work, including the decision to publish.
Author information
Authors and Affiliations
Contributions
AM, AD and GO wrote the manuscript; RAK, PK, MB, CN, SB and SR designed the study and edited the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This project has been approved by the Human Research Ethics Committees of Austin Health (approval 16/Austin/188) and The Melbourne Clinic (approval 305). All participants will give written informed consent.
Consent for publication
Not applicable.
Competing interests
MB is supported by a NHMRC Senior Principal Research Fellowship (1059660 and 1156072).
MB has received Grant/Research Support from the NIH, Cooperative Research Centre, Simons Autism Foundation, Cancer Council of Victoria, Stanley Medical Research Foundation, Medical Benefits Fund, National Health and Medical Research Council, Medical Research Futures Fund, Beyond Blue, Rotary Health, A2 milk company, Meat and Livestock Board, Woolworths, Avant and the Harry Windsor Foundation, has been a speaker for Astra Zeneca, Lundbeck, Merck, Pfizer, and served as a consultant to Allergan, Astra Zeneca, Bioadvantex, Bionomics, Collaborative Medicinal Development, Lundbeck Merck, Pfizer and Servier – all unrelated to this work.
CN has served as a consultant for Lundbeck, Grunbiotics, Servier, Janssen-Cilag, Wyeth and Eli Lilly, received research grant support from Wyeth and Lundbeck, and speaker honoraria from Servier, Lundbeck, Bristol-Myers Squibb, Organon, Eli Lilly, GlaxoSmithKline, Janssen- Cilag, Astra-Zenaca, Wyeth, and Pfizer.
The other authors declare they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Maier, A., Dharan, A., Oliver, G. et al. A multi-centre, double-blind, 12-week, randomized, placebo-controlled trial to assess the efficacy of adjunctive N-Acetylcysteine for treatment-resistant PTSD: a study protocol. BMC Psychiatry 20, 397 (2020). https://doi.org/10.1186/s12888-020-02793-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12888-020-02793-9