
Abstract
Background
The evidence on selective serotonin reuptake inhibitors (SSRIs) for major depressive disorder is unclear.
Methods
Our objective was to conduct a systematic review assessing the effects of SSRIs versus placebo, ‘active’ placebo, or no intervention in adult participants with major depressive disorder. We searched for eligible randomised clinical trials in The Cochrane Library’s CENTRAL, PubMed, EMBASE, PsycLIT, PsycINFO, Science Citation Index Expanded, clinical trial registers of Europe and USA, websites of pharmaceutical companies, the U.S. Food and Drug Administration (FDA), and the European Medicines Agency until January 2016. All data were extracted by at least two independent investigators. We used Cochrane systematic review methodology, Trial Sequential Analysis, and calculation of Bayes factor. An eight-step procedure was followed to assess if thresholds for statistical and clinical significance were crossed. Primary outcomes were reduction of depressive symptoms, remission, and adverse events. Secondary outcomes were suicides, suicide attempts, suicide ideation, and quality of life.
Results
A total of 131 randomised placebo-controlled trials enrolling a total of 27,422 participants were included. None of the trials used ‘active’ placebo or no intervention as control intervention. All trials had high risk of bias. SSRIs significantly reduced the Hamilton Depression Rating Scale (HDRS) at end of treatment (mean difference −1.94 HDRS points; 95% CI −2.50 to −1.37; P < 0.00001; 49 trials; Trial Sequential Analysis-adjusted CI −2.70 to −1.18); Bayes factor below predefined threshold (2.01*10−23). The effect estimate, however, was below our predefined threshold for clinical significance of 3 HDRS points. SSRIs significantly decreased the risk of no remission (RR 0.88; 95% CI 0.84 to 0.91; P < 0.00001; 34 trials; Trial Sequential Analysis adjusted CI 0.83 to 0.92); Bayes factor (1426.81) did not confirm the effect). SSRIs significantly increased the risks of serious adverse events (OR 1.37; 95% CI 1.08 to 1.75; P = 0.009; 44 trials; Trial Sequential Analysis-adjusted CI 1.03 to 1.89). This corresponds to 31/1000 SSRI participants will experience a serious adverse event compared with 22/1000 control participants. SSRIs also significantly increased the number of non-serious adverse events. There were almost no data on suicidal behaviour, quality of life, and long-term effects.
Conclusions
SSRIs might have statistically significant effects on depressive symptoms, but all trials were at high risk of bias and the clinical significance seems questionable. SSRIs significantly increase the risk of both serious and non-serious adverse events. The potential small beneficial effects seem to be outweighed by harmful effects.
Systematic review registration
PROSPERO CRD42013004420.
Similar content being viewed by others


Background
Selective serotonin reuptake inhibitors (SSRIs) are often first-line treatment for depression and prescriptions for SSRIs are increasing [1, 2]. A number of reviews with meta-analysis have assessed the effects of SSRIs in adults with major depressive disorder [3,4,5,6,7,8], generally concluding that SSRIs have a statistically significant effect on depressive symptoms [3,4,5,6,7,8]. However, the results of the reviews have been limited by not using predefined Cochrane methodology [3,4,5,6,7,8], only including subgroups of depressed patients [9, 10], not searching all relevant databases [3,4,5,6,7,8, 10], not systematically assessing harms [3,4,5,6,7,8, 10], and not systematically assessing risks of bias [3,4,5,6,7,8, 10]. We have summarised the characteristics and the results of previous systematic reviews in Table 1. Accordingly, the evidence on the effects of SSRIs is unclear. Using, e.g., a composite outcome of all serious adverse events (according to ICH-GCP [11]) might show how SSRIs work. Furthermore, assessments of quality of life might demonstrate if SSRIs have clinically meaningful effects. It is of utmost importance to assess the clinical significance of review results if statistically significant results are shown [12, 13].
Our objective was to conduct a comprehensive systematic review assessing the beneficial and harmful effects of SSRIs versus placebo, ‘active’ placebo, or no intervention in adult participants with major depressive disorder using our eight-step procedure for assessing evidence in systematic reviews [13].
Methods
Details regarding the methodology are described in our protocol, which was registered prior to the systematic literature searches [14]. The methodology was not changed after the analysis of the review results began [14].
We included all randomised clinical trials comparing the effects of SSRIs (citalopram; escitalopram; sertraline; fluoxetine; paroxetine; or fluvoxamine) versus placebo, ‘active placebo’ (any active substance employed to mimic the adverse effects of taking a SSRI) [15], or no intervention. We also planned to perform subgroup analyses comparing the effects of the different doses (see Subgroup analyses). If a trial had three arms (e.g., a three-arm trial randomising the participants to two different SSRIs and placebo) then we divided the total number of control participants with two but kept the means and SDs in each group unchanged [16, 17].
Independent investigators searched for eligible trials published before January 2016 in The Cochrane Library’s CENTRAL, PubMed, EMBASE, PsychLIT, PsycINFO, clinicaltrials.gov., and Science Citation Index Expanded [14] (see Additional file 1: Search strategies). Trials were included irrespective of language, publication status, publication year, and publication type. To identify unpublished trials, we searched clinical trial registers of Europe and USA, websites of pharmaceutical companies, websites of U.S. Food and Drug Administration (FDA) and European Medicines Agency, and we requested the U.S. Food and Drug Administration (FDA) to provide all publicly releasable information about relevant clinical trials of SSRIs that were submitted for marketing approval.
Participants had to be 18 years or older and have a primary diagnosis of major depressive disorder based on standardised criteria, such as DSM III, DSM III-R, DSM IV, DSM V, or ICD 10 [14].
We excluded trials specifically randomising depressed participants with: a specific somatic disease, schizophrenia, or depression during or after pregnancy.
Outcomes
Primary outcomes
-
Depressive symptoms measured on the 17-item or 21-item Hamilton Depression Rating Scale (HDRS) [18], the Montgomery-Asberg Depression Rating Scale (MADRS) [19], or the Beck’s Depression Inventory (BDI) [20].
-
Remission (Hamilton <8 points; BDI <10 points; MADRS <10 points).
-
Adverse events during the intervention period which were classified as serious and non-serious adverse events [11]. Serious adverse events were defined as medical events that were life threatening, resulted in death, disability, or significant loss of function, or caused hospital admission or prolonged hospitalisation [11]. The remaining events were classified as non-serious adverse events [11].
Secondary outcomes
-
Suicides, suicide attempts, and suicide ideation during the intervention period.
-
Quality of life (scale used by the trialists).
The time point of primary interest was end of treatment (defined by trialist) [14]. We also planned to report results assessed at maximum follow-up [14].
Handling of missing data
If the participant was not included in the analysis of ‘no remission’, we assumed that they had ‘no remission’ [17]. If, e.g. 23/50 participants had ‘no remission’ but a total of 53 were randomised then we assumed that 26 had ‘no remission’. For all remaining outcomes we used observed data when these were reported [17].
Subgroup analyses
We planned the following subgroup analyses: participants with HDRS baseline ≤23 compared to >23 points; different SSRIs; trials using a placebo washout period before randomisation compared to trials using no washout period; participants with alcohol or drug dependence compared to no dependence; elderly participants compared to younger participants; duration of treatment below 8 weeks compared to equal to or above 8 weeks; and SSRI below or equal to median dose compared to above median dose.
We planned to assess if SSRIs differ according to psychiatric comorbidities [17]. Four trials included depressed participants with comorbid anxiety but none of these trials reported HDRS so this could not be performed. No other comorbidities (including borderline personality disorder, chronic depression, and treatment resistant depression) were identified in the included trials. We planned to assess if the effects of SSRIs differed: (1) when the SSRI was delivered as add-on therapy to another antidepressant drug; (2) per use of different forms of control interventions (‘active placebo’, traditional placebo, and no intervention); and (3) if electroconvulsive therapy was used as co-intervention. However, these analyses were not possible.
We used test for subgroup differences to assess if the effects of SSRIs seemed to differ between the different types of participants if either a trial specifically randomised a certain type of participants, or a trial reported results separately for each specific type of participants.
Selection of trials and data extraction
Review authors (KKK, AS, SGH, SES, KLM, MI, MBB, IJP, JK, SLK, AT, SE, JCJ) worked in pairs and independently selected relevant trials and extracted data. A standardised data extraction sheet was used (see Data extraction form). If a trial was identified by only one, it was discussed whether the trial should be included. In case of discrepancy, a third review author (JCJ) was consulted. We contacted review authors if relevant information was missing.
Assessment of the statistical and clinical significance
Our methodology was based on The Cochrane Handbook and GRADE [14, 16, 21]. We assessed statistical and clinical significance according to our eight-step procedure [13]:
-
(1)
We obtained 95% confidence intervals (95% CI) and P-values from all planned random-effects [22] and fixed-effect meta-analyses [16] and reported the most conservative result as the main result [13]. Review Manager version 5.3 was used for all meta-analyses [23].
-
(2)
Sensitivity analyses and subgroup analyses were conducted to explore the reasons for substantial statistical heterogeneity [13, 16]. Statistical heterogeneity was assessed by visual inspection of forest plots and by the heterogeneity (I2 or D2) [13, 16, 24, 25].
-
(3)
We defined three primary outcomes in our protocol [14]. Our threshold for significance was therefore adjusted according to problems with multiplicity [13] by dividing 0.05 with the value halfway between 1 (no adjustment) and 3 (Bonferroni adjustment) [13, 17] resulting in 0.05/2 = 0.025.
-
(4)
Cumulative meta-analyses are at risk of producing random errors due to sparse data and multiple testing of accumulating data [25, 26]. Therefore, Trial Sequential Analysis version 0.9.5.5 beta was applied to control this risk (http://www.ctu.dk/tsa/) [27]. The required information size (that is the number of participants needed in a meta-analysis to detect or reject a certain intervention effect) was calculated [25, 28]. The required information size is based upon the event proportion in the control group; the assumption of a plausible relative risk (RR) reduction; and the assumed heterogeneity or diversity of the meta-analysis [25, 29]. Trial Sequential Analysis enables testing to be conducted each time a new trial is included in the meta-analysis. Based on the required information size, trial sequential monitoring boundaries are constructed. This enables one to determine the statistical inference concerning cumulative meta-analysis that has not yet reached the required information size [25]. Firm evidence may be established if one of the trial sequential monitoring boundaries (for benefit (upper red dotted line), for harm (lower red dotted line), or for futility (vertical red dotted line)) is crossed by the cumulative Z-score before reaching the required information size, in which case further trials may turn out to be superfluous [28]. Trial Sequential Analysis- adjusted confidence intervals are also presented [28]. For dichotomous outcomes, we estimated the diversity-adjusted required information size based on the proportion of patients with an outcome in the control group, a relative risk reduction of 30%, an alpha of 2.5%, a beta of 20%, and diversity in the trials [13, 17]. For continuous outcomes, we estimated the required information size based on a HDRS mean difference of three points, the standard deviation observed in the control group, an alpha of 2.5%, a beta of 20%, and diversity in the trials [13, 17]. All outcomes were assessed with Trial Sequential Analysis [13, 24, 25].
-
(5)
We calculated Bayes factors for all primary outcomes. A low P value indicates that an observed result is unlikely given that there is no difference in effect between the compared intervention groups (i.e., the null hypothesis is true) [12, 13]. Even very low P values may, therefore, be misleading because the probability that the actual measured difference in effect of the compared interventions resulted from an a priori anticipated ‘true’ difference needs to be considered [13]. For this purpose, it is helpful to calculate Bayes factor for the primary outcomes. It will show the ratios between the P-value probabilities of the meta-analysis result divided by the probability of the meta-analysis result given that an anticipated intervention effect is the true effect [12, 13]. In other words, the lower the Bayes factor gets the more confident one should be that an actual intervention effect (the anticipated intervention effect) has produced the meta-analysis results and that a given difference between the compared groups is not caused by random error [12, 13]. Calculation of Bayes factor is not part of standard Cochrane methodology.
-
(6)
We assessed the potential impact of bias on the review results [16]. To assess the potential impact of missing data (incomplete outcome data bias) we assessed a ‘best-worst’ case scenario assuming that all participants lost to follow-up in the SSRI group had a beneficial outcome (the group mean plus 1 standard deviation (SD) or plus 2 SDs); and all those with missing outcomes in the placebo group have had a harmful outcome (the group mean minus 1 SD or minus 2 SDs) [13, 17]. We also performed the reverse ’worst-best-case’ scenario analysis [13, 17].
-
(7)
We assessed the risk of publication bias by visual inspection of funnel plots and by tests for funnel plot asymmetry [13, 16, 30].
-
(8)
We assessed clinical significance of our results. As previously suggested [4, 8, 31], we chose a drug-placebo difference of 3 points on the 17-item HDRS or an effect size of 0.50 SMD as the threshold for clinical significance (see Discussion) [14].
Results
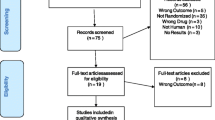
We have summarised the selection of trials in Fig. 1 and excluded trials in Additional file 2: List of excluded trials.

PRISMA flowchart
Using our strict inclusion and exclusion criteria, a total of 195 publications/unpublished trials were identified and included. Due to multiple publications of single trials and lack of useful data, only 131 trials randomised clinical trials [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164] enrolling a total of 27,422 participants were included in our analyses. 116 were published trials [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,121,122,123,124,125,126,127,128,129,130,134,135,147,157,158,159,160,161,162,163,118, 120–131, 133–136, 141, 144, 146–148, 152, 153, 156–164] and 15 were unpublished trials [102,138,139,150,, 119, 132, 137–140, 142, 143, 145, 149–151, 155, 165]. Of the 15 unpublished trials, eight were identified via company websites, two via clinicaltrials.gov, and four via FDA (see Additional file 3: Trial Characteristics).
We did not identify any trials using ‘active placebo’ or ‘no intervention’ as control interventions. Most trials used broad inclusion criteria and randomised adult depressed women and men below 65 years and used SSRI for 6 to 12 weeks. Twelve of the included trials specifically randomised elderly (most often participants >65 years) depressed participants [45, 48, 54, 63, 82, 110, 111, 113, 122, 144, 162, 166], five trials randomised depressed alcohol dependant participants [47, 61, 116, 120, 156], and three trials randomised depressed opioid and cocaine dependant participants [44, 115, 164]. Types of the randomised participants and other trial characteristics are summarised in Additional file 3: Trial characteristics.
Primary outcomes
Hamilton depression rating scale (HDRS)
Twenty-two trials reported mean HDRS scores and standard deviation (SD) [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50, 103, 144, 162] and 27 trials reported mean HDRS change scores and SD at end of treatment [51,55,56,57,58,59,60,61,62,63,64,65,, 52, 54–66, 121, 123, 137, 138, 141, 142, 145, 146, 167, 168]. Random-effects meta-analysis of these 49 trials showed that SSRIs versus placebo significantly reduced the HDRS score (mean difference −1.94 points; 95% CI −2.50 to −1.37; P < 0.00001) (Fig. 2). Twenty-four trials reported only mean HDRS scores or presented a graph showing the mean HDRS scores, but did not report the SD at end of treatment [67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88, 166]. We planned to impute missing SDs based on observed standard deviations from trials with similar characteristics [17]. Trial characteristics, sample sizes, and statistical weight of the included trials were similar across trials and we therefore chose to impute the missing SDs with a value of 8 points (the mean of the observed standard deviations rounded up to the nearest integer). Nineteen trials reported only mean HDRS change scores or presented a graph showing the mean change HDRS scores, but did not report the SD [89,90,91,92,93,94,95,96,97,98,99,100,101,102, 108, 143, 157, 158, 169]. We imputed the missing SDs with a value of 7 points (the mean of the observed standard deviations rounded up to the nearest integer) [17]. Random-effects meta-analysis of the results of all 92 trials showed that SSRIs versus placebo significantly reduced the HDRS score (mean difference −2.25 points; 95% CI −2.69 to −1.83; P < 0.00001).

Random-effects meta-analysis of the results on the Hamilton depression rating scale
Heterogeneity
The primary meta-analysis showed statistically significant heterogeneity (I 2 = 75%; P < 0.00001) (Fig. 2). Visual inspection of the forest plot indicated that one trial seemed to have a more extreme effect (larger intervention effect estimate and smaller confidence interval) [58]. For exploratory purposes, we tried to exclude this trial from the analysis and this reduced the I2 to 29%, but removing this trial did not substantially alter the meta-analysis result (mean difference after removing the trial from the analysis −1.77 HDRS points; 95% CI −2.12 to −1.42).
Subgroup analysis per risk of bias
All the included trials had high risk of bias. Hence, it was not possible to perform a subgroup analysis of trials at low risk of bias (Fig. 3) [13, 17]. However, four trials [33, 60, 103, 121] were classified as potentially lower risk of bias (based on bias risk assessment of ‘generating allocation sequence’, ‘allocation concealment’, ‘blinding of participants and treatment providers’, and ‘blinding of outcome assessment’) [13, 17]. Random-effects meta-analysis of these four trials showed that SSRIs versus placebo reduced the HDRS score (mean difference −2.07 points; 95% CI −3.06 to −1.08). Test for subgroup differences between trials with lower risk of bias (n = 4) compared to trials with high risk of bias (n = 45) was not significant (P = 0.82).

Risk of bias in the included randomized clinical trials
Incomplete outcome data
Meta-analysis of the best-worst case scenario analyses adding 1 SD (mean difference −3.38 HDRS points in favour of SSRI; 95% CI −4.10 to −2.66) and adding 2 SDs (mean difference −4.50 HDRS points in favour of SSRI; 95% CI −5.37 to −3.63) for missing values showed large significant intervention effect estimates [13, 17]. Meta-analysis of the worst-best case scenario analyses showed a significant intervention effect estimate when adding 1 SD for missing values (mean difference −0.77 points; 95% CI −1.45 to −0.09) and no significant intervention effects when adding 2 SDs for missing values (mean difference 0.46 points; 95% CI −0.38 to 1.30).
Other subgroup analyses
Meta-analysis of the results of the 26 trials with a mean baseline HDRS score >23 points showed a mean difference of −2.69 HDRS points; 95% CI −3.59 to −1.78; P < 0.00001 [32,36,37,46,58,64,, 35–38, 41, 43, 45–47, 49, 52, 57–59, 63–65, 121, 137, 142, 145, 167, 170, 171]. Meta-analysis of the results of the 20 trials with a mean baseline HDRS score ≤23 points showed a mean difference of -1.29 HDRS points; 95% CI −1.76 to −0.82; P < 0.00001 [33,61,62,, 34, 39, 40, 44, 48, 50, 51, 54, 55, 60–63, 103, 141, 144, 146, 162, 168]. Test for subgroup difference was significant (I 2 = 86.2%; P = 0.007). We performed post-hoc meta-regression (STATA 14) with baseline HDRS as a covariate in the meta-analysis. This analysis showed that the effects of SSRIs seem to increase with larger baseline HDRS scores (coefficient −0.33 points; 95% CI −0.44 to −0.22; P < 0.0001).
The following tests for subgroup differences did not show any significant differences: trials assessing the effects of the different SSRIs (number of trials 49; I 2 = 2.2%; P = 0.40) (Fig. 4); published trials (45 trials) compared to unpublished trials (4 trials): I 2 = 25.2%; P = 0.25; trials randomising elderly participants (6 trials) compared to younger participants (43 trials): I 2 = 0%; P = 0.94 (Fig. 5); trials with washout period (40 trials) compared to trials without washout period (9 trials): I 2 = 63.6%; P = 0.10 (Fig. 6); trials randomising drug or alcohol dependant participants (3 trials) compared to the remaining trials (46 trials): I 2 = 0%; P = 0.58; trials with an intervention period below 8 weeks (19 trials) compared to the remaining trials (30 trials): I 2 = 36.6; P = 0.21; and dose of the chosen SSRI (dose below the median (6 trials) compared to equal to or above the median (9 trials) of the SSRI: I 2 = 0%; P = 0.65. The latter two subgroup analyses were post-hoc analyses.

Subgroup analysis comparing trials assessing the effects of different selective serotonin reuptake inhibitors

Subgroup analysis comparing trials randomising elderly participants to trials randomising non-elderly participants

Subgroup analysis comparing trials using a placebo-washout period to trials not using a placebo-washout period
We performed an additional post-hoc subgroup analysis comparing trials with low risk of financial bias to trials with high risk of financial bias (Additional file 4: Figure S1). Test for subgroup differences showed no significant difference (P = 0.18). When the four trials with low risk of bias of financial bias were analysed separately then there was no significant difference between the SSRI group and the placebo group (−0.92 points; 95% CI −2.42 to 0.58; I2 26%) (Additional file 4: Figure S1).
Trial Sequential Analysis
The required information size was calculated based on an anticipated intervention effect of 3 HDRS points, the empirical variance, a risk of type I error of 0.025, and a power of 80% [12, 13, 17]. The Trial Sequential Analysis showed that the trial monitoring boundary for benefit was crossed after the 9th trial indicating a statistically significant result (Trial Sequential Analysis-adjusted CI −2.62 to −1.26) (Fig. 7).

Trial Sequential Analysis of the results of selective serotonin reuptake inhibitors on Hamilton depression rating scale
Bayes factor
Bayes factor was calculated based on our anticipated intervention effect of 3 HDRS points and the primary meta-analysis result (mean difference −1.94 points; 95% CI −2.50 to −1.37) [12, 13, 17]. Bayes factor (2.01*10−23) was below the threshold for significance of 0.1, supporting the statistical significant result.
Risk of publication bias
Visual inspection of the funnel plot did not show clear signs of asymmetry (Fig. 8) [13, 17].

Funnel plot of the random-effects meta-analysis of the effect of selective serotonin reuptake inhibitors on Hamilton depression rating scale
Hamilton Depression Rating Scale, Montgomery-Asberg Depression Rating Scale, and Beck’s Depression Inventory
Sixty-one trials [32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,55,56,57,58,59,60,61,62,63,64,65,104,105,138,139,140,141,145,150,52, 54–66, 103–106, 120, 121, 123, 137–142, 144–146, 149–151, 154, 155, 168, 172, 173] reported means and SD on the HDRS [18], MADRS [19], or BDI [174]. Using standardised mean difference, random-effects meta-analysis showed that SSRI versus placebo significantly decreased the standardised mean difference score (trials reporting mean scores: −0.23; 95% CI −0.31 to −0.14; P < 0.00001; trials reporting mean change scores: −0.26; 95% CI −0.35 to −0.17; P < 0.00001). The standardised mean difference was below our predefined threshold for clinical significance.
Long-term follow-up
One trial [33] reported mean HDRS scores and SD and one trial [146] reported mean HDRS change scores and SD at end of long-term follow-up. Random-effects meta-analysis of these trials showed a mean difference −0.18 points (95% CI −2.78 to 2.43; P = 0.89). Four trials reported mean HDRS scores or change scores at end of long-term follow-up but without reporting SDs [70, 97, 107, 143]. SDs were imputed. Random-effects meta-analysis of all the six trials showed a mean difference of −1.30 points (95% CI −2.72 to 0.13; P = 0.07).
No remission
Thirty-four trials [33,50,51,52,108,109,110,111,, 34, 38, 45, 49–53, 55, 56, 60, 78, 81, 101, 104, 107–112, 120, 128, 140, 141, 146, 153, 157, 158, 162, 171, 175, 176] reported the proportion of participants with no remission at end of treatment. A total of 1430/2211 (64.7%) SSRI participants experienced no remission compared with 1493/2003 (74.5%) control participants. Random-effects meta-analysis showed that SSRIs versus placebo significantly decreased the risk of no remission (RR 0.88; 95% CI 0.84 to 0.91; P < 0.00001) (Fig. 9). This corresponds to 657 (95% CI 642 to 679) SSRI participants out of 1000 will experience no remission compared with 746 control participants out of 1000 (see Additional file 5: Summary of findings table). Visual inspection of the forest plots showed no clear signs of heterogeneity [13, 16].

Random-effects meta-analysis of the results of selective serotonin reuptake inhibitors on remission of depression
The required information size was calculated based on the observed proportion of control participants without remission, a relative risk reduction of 30%, a risk of type I error of 0.025, and a power of 80% [13, 17]. The Trial Sequential Analysis showed that the trial monitoring boundary for benefit was crossed (Fig. 10) and the Trial Sequential Analysis-adjusted CI was 0.83 to 0.92 [13, 17].

Trial Sequential Analysis of the results of selective serotonin reuptake inhibitors on remission of depression
Bayes factor was 1426.8 based on the random-effects meta-analysis result and above the threshold for significance of 0.1 [13, 17]. This relatively high Bayes factor indicates that it is more likely that null effect (null hypothesis) compared to the anticipated intervention effect has produced this meta-analysis results [12, 13].
Best-worst case scenario showed a highly significant meta-analysis result (RR 0.78; 95% CI 0.73 to 0.83; P < 0.00001) [13, 17]. Worst-best case scenario showed no significant difference on risk of no remission (RR 0.95; 95% CI 0.89 to 1.02; P = 0.14) [13, 17].
Visual inspection of the funnel plot showed no clear signs of publication bias [13, 16, 17].
Serious adverse events
Because of the low proportion in the control group (around 2%) we used the Mantel–Haenszel (MH) odds ratio method with reciprocal zero cell correction (zero is replaced by the reciprocal of the size of the opposite treatment arm) [177]. Forty-four trials reported the proportion of participants with serious adverse events [48,55,113,114,115,116,117,138,145,150,179,, 49, 54–56, 60, 63, 75, 78, 93, 94, 102, 105, 108, 112–118, 120, 121, 137–139, 141, 144–146, 149–151, 155, 167, 171, 173, 176, 178–180]. A total of 239/8242 (2.7%) SSRI participants experienced a serious adverse event compared with 106/4956 (2.1%) control participants. Random-effects meta-analysis showed that SSRIs versus placebo significantly increased the risk of a serious adverse event (OR 1.37; 95% CI 1.08 to 1.75; P = 0.009) (Fig. 11). This corresponds to 31 (95% CI 25 to 40)/1000 SSRI participants will experience a serious adverse event compared with 22/1000 control participants (see Additional file 5: Summary of findings table). Visual inspection of the forest plot did not indicate significant heterogeneity [13, 17]. Even when using the multiplicity adjusted risk of type I error (0.05/2 = 0.025), the trial sequential boundary for harm was crossed (Fig. 12) and the Trial Sequential Analysis-adjusted CI was 1.03 to 1.89 [13, 17]. Table 2 summarises the types of adverse events.

Random-effects meta-analysis of the results of selective serotonin reuptake inhibitors on serious adverse events

Trial Sequential Analysis of the results of selective serotonin reuptake inhibitors on serious adverse events
Bayes factor was 4.8*105 above the threshold for significance of 0.1 [13, 17]. This clearly shows that a beneficial effect of SSRIs on serious adverse events is very unlikely [13, 17]. Visual inspection of the funnel plot showed no signs of publication bias [13, 16, 17]. Based on the random-effects meta-analysis result, we calculated the number-needed-to-seriously harm one patient to be 138 patients.
Adverse events
Meta-analyses showed that the participants randomised to SSRIs versus placebo had a significantly increased risk of several adverse events. We have summarised the risks of the adverse events which were most reported in Additional file 6, including numbers-needed-to-harm. We have also included forest plots for the 25 most statistically significant adverse event results in the Additional files (see Additional files 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30 and 31: Figure S3-S27). The full list of the 84 reported adverse events are summarised in Table 3.
Clinical significance
All primary HDRS meta-analyses showed intervention effect estimates below our predefined threshold for clinical significance (a mean difference of 3 HDRS points or 0.5 standardised mean difference) [13, 17]. Our results show statistically significant effects, but the possible effects all seem to have questionable clinical significance [13].
Secondary outcomes
Suicides, suicide attempts, and suicide ideation
There were no significant differences between participants randomised to SSRIs versus placebo on number of suicides (RR 0.68; 95% CI 0.16 to 2.81; P = 0.59; Trial Sequential Analysis-adjusted CI 0.01 to 226.85; 6 trials [60, 71, 108, 113, 151, 155]); suicide attempts (RR 1.76; 95% CI 0.59 to 5.22; P = 0.31; Trial Sequential Analysis-adjusted CI 0.02 to 149.95; 8 trials [49, 56, 75, 94, 102, 139, 167, 181]); or suicide ideation (RR 0.80; 95% CI 0.36 to 1.77; P = 0.58; Trial Sequential Analysis-adjusted CI 0.03 to 23.20; 11 trials [49, 51, 120, 138, 139, 145, 151, 162, 167, 171, 180]). The required information size was not reached in any of the three Trial Sequential Analyses.
Quality of life
Only six trials assessed quality of life [48, 51, 63, 100, 101, 112] out of which four trials reported results on the quality of life enjoyment and satisfaction questionnaire (Q-LES-Q) [48, 51, 100, 101]. Two trials [48, 101] reported mean scores and SDs. Random-effects meta-analysis showed significant effect of SSRI on Q-LES-Q scores (RR 2.98; 95% CI 1.34 to 4.61; P = 0.0004).
Two trials reported results on the short form of the quality of life enjoyment and satisfaction questionnaire [63, 112], but only one trial reported mean scores and SDs [63]. The results from this trial showed that SSRIs (paroxetine) versus placebo significantly increased the mean score of the questionnaire (paroxetine 12.5 mg group mean 11.4, SD 16.7; paroxetine 25 mg group mean 11.5, SD 17.2; placebo group mean 5.3, SD 17.1) [63].
Post hoc analysis of no response
We identified 70 trials assessing the effects of SSRIs on no response defined as less than 50% reduction (from baseline) on either HDRS or MADRS. The meta-analysis showed that SSRIs seem to significantly decrease the risk of no response compared with placebo (RR 0.83; 95% CI 0.80 to 0.87; P = 0.00001) (Additional file 32: Figure S2).
GRADE assessments
GRADE assessments show that due to the high risks of bias the quality of the evidence must be regarded as very low (Additional file 5: Summary of Findings Table) [13].
Discussion
SSRIs may affect the concentration of essential neurotransmitter substances in the brain and are therefore considered to exert effects on depressive symptoms. However, whether these effects are beneficial and clinically meaningful are the questions. Estimating a meaningful threshold for clinical significance is difficult and an assessment of clinical significance should ideally not only include a threshold on an assessment scale [182]. Major depressive disorder affects daily functioning, increases the risk of suicidal behaviour, and decreases quality of life [183]. Some adverse events might therefore be acceptable if SSRIs have clinically significant beneficial effects [13, 183, 184]. We therefore both predefined a threshold for clinical significance and assessed the balance between beneficial and harmful effects [13, 17, 184].
As threshold for clinical significance [14], we chose a drug-placebo difference of 3 points on the 17-item HDRS (ranging from 0 to 52 points) or an effect size of 0.50 standardised mean difference. This has been recommended by the National Institute for Clinical Excellence (NICE) in England and has been chosen in other reviews [4, 8, 31]. Nevertheless, these recommendations are not universally accepted and have been questioned [3]. Others have suggested the following ‘rules of thumb’ regarding the standardised mean difference: 0.2 a small effect, 0.5 a moderate effect, and 0.8 a large effect [16, 185]. One study has shown that a SSRI-placebo mean difference of up to three points on the HDRS corresponds to ‘no clinical change’ [186]. Another valid study has shown that a SSRI-placebo difference of 3 points is undetectable by clinicians, and that a mean difference of 7 HDRS points, or a standardized mean effect size of 0.875, is required to correspond to a rating of ‘minimal improvement’ [187]. It has been speculated that the ‘placebo’ response in antidepressant trials has been increasing during recent years [188]. If there is a ‘response’ to placebo this has of course to be considered when interpreting a mean difference between drug and placebo. However, it is unlikely that depressed patient have a significant placebo effect [189] and it has recently been shown that the placebo response has been stable for 25 years [188]. Even based on our predefined minimal thresholds for clinical significance, the effects of SSRIs did not have a clinically meaningful effect on depressive symptoms. Furthermore, per our meta-analyses SSRIs significantly increase the risk of both serious and non-serious adverse events.
The best-worst and worst-best case scenarios showed that incomplete outcome data bias alone theoretically could have caused the apparent statistically significant beneficial effect of SSRIs. Furthermore, seen in the light of the total number of trials, only a relatively limited number of trials reported on each of our pre-defined outcomes. This increases the risk of selective outcome reporting bias. Apart from the high risk of incomplete outcome data bias and selective outcome reporting bias, all the included trials were assessed at high risk of bias. All trials used placebo as control intervention and due to the large number of adverse events, some patients might have figured out if they received an ‘active’ intervention or not, which might question the blinding of the trials. Nevertheless, it may be argued that our bias risk assessment often will lead to no trials with low risk of bias. However, similar bias risk assessments have been used in several previous systematic review (see, e.g., most Cochrane Hepato-Biliary Group systematic reviews) and our bias risk assessment is based on valid evidence clearly showing that if each of the used bias risk domains is ‘high risk of bias’ or ‘unclear risk of bias’ then there is a risk of overestimation of benefits and underestimation of harms [184,191,192,193,194,195,196,, 190–197]. Furthermore, the risks of bias observed here just mirrors our experience in 786 randomised trials on depression [198].
We chose ‘remission’ as a primary outcome because we expected trialists to use this outcome frequently. To present a complete overview of the evidence on SSRIs for depression we also included ‘no response’ (less than 50% reduction on HDRS or MADRS during the intervention period) in a post hoc analysis because this outcome was frequently used in the included trials and by requests from peer reviewers. However, our results on no remission and no response should be interpreted with great caution for a number of reasons: 1) the assessments of remission and response were primarily based on single HDRS scores and it is questionable whether single HDRS scores are indications of full remission or adequate response to the intervention; 2) information is lost when continuous data are transformed to dichotomous data and the analysis results can be greatly influenced by the distribution of data and the choice of an arbitrary cut-point [16,200,, 199–201]; 3) even though a larger proportion of participants cross the arbitrary cut-point in the SSRI group compared with the control group (often HDRS below 8 for remission and 50% HDRS reduction for response), the effect measured on HDRS might still be limited to a few HDRS points (e.g., 3 HDRS points) or less; 4) by only focusing on how many patients cross a certain line for benefit, investigators ignore how many patients are deteriorating at the same time. If results, e.g., show relatively large beneficial effects of SSRIs when remission and response are assessed but very small averaged effects (as our results show) – then it must be because similar proportions of the participants are harmed (increase on the HDRS compared to placebo) by SSRIs. Otherwise the averaged effect would not show small or no difference in effect. The clinical significance of our results on ‘no remission’ and ‘no response’ should therefore be questioned. The methodological limitations of using ‘response’ as an outcome has been investigated in a valid study by Kirsch et al. who conclude that: “response rates based on continuous data do not add information, and they can create an illusion of clinical effectiveness” [202]. In retrospect, due to these methodological limitations we should not have assessed ‘no remission’ or ‘no response’ as outcomes. This is a clear limitation of our review [16,200,, 199–201].
Our tests for subgroup difference comparing trials with a baseline HDRS score below and above 23 points and meta-regression showed that the effects of SSRIs seem to increase with increased baseline HDRS score. Others have also shown that trials randomising participants with a higher baseline HDRS mean average seem to show larger effects of antidepressants [7, 8]. However, it is difficult to interpret why trials with higher average baseline HDRS score seem to have a larger effect of SSRIs. This might just be due to random error. No matter, it cannot be concluded based on these results that SSRIs work better on more severely depressed patients. To make such a conclusion individual patient data would be necessary, i.e., it would be necessary to show that it is actually the patients with higher baseline HDRS scores who have the larger effects. Gibbons et al. used longitudinal person-level data from a large set of published and unpublished studies and showed baseline severity was not significantly related to degree of SSRI treatment advantage over placebo [3]. It must be noted that the intervention effects in the group with HDRS scores above 23 points were still below our threshold for clinical significance, supporting Gibbons and co-workers’ results.
Leucht et al. have suggested that effects sizes of SSRIs in randomised clinical trials have declined over time [203]. Post-hoc meta-regression of the HDRS results confirmed their results (effect sizes going down from around 0.8 in the early 1980s to 0.25 in 2012). The reasons for the decreasing effect is not entirely understood but might be due to better methodology nowadays or recruitment of different types of participants [203]. Leucht et al. also suggested that a lack of difference between antidepressants and placebo is caused by an increasing ‘placebo’ effect (spontaneous recovery) [203]. This seem less important from a patient perspective, i.e., whether a certain drug should be used should be based on the benefits and harms of this drug compared with placebo. Furthermore, the increasing placebo effect has recently been severely questioned [188].
Our present systematic review has several strengths. Our protocol was registered prior to the systematic literature search in all relevant databases, data extraction, and data analyses [14]. Data were double-extracted by independent authors minimising the risk of inaccurate data extraction, and we assessed the risk of bias in all trials according to Cochrane [16]. We used Trial Sequential Analysis to control the risks of random errors [25, 29, 204], and the analyses of the primary outcomes showed that the accrued information sizes were sufficient. Both visual assessments of forest plots and statistical test showed limited signs of statistical heterogeneity, e.g., I2 was 0% when assessing risk of serious adverse events. Hence, these findings increase the validity of our review results and indicate that the effects shown are consistent across the different trials. Multiple previous reviews and meta-analyses have, as mentioned in our Background, assessed the effects of SSRIs and have generally concluded that SSRIs have significant effects on depressive symptoms [3,4,5,6,7,8]. However, the estimated results (and not the conclusions the review authors made) of these reviews and meta-analyses actually are in agreement with our present results and show that SSRIs do not seem to benefit patients more than a few HDRS points. This increases the validity of our present results. Furthermore, we assessed in detail the risks of serious adverse events and of non-serious adverse events and found that both were significantly increased by SSRIs.
Our systematic review has several limitations. Our HDRS mean differences were averaged effects. Hence, it cannot be concluded that SSRIs do not have clinically significant effects on all depressed participant. E.g., certain severely depressed patients compared with lightly depressed patients (e.g., so-called professional patients or symptomatic volunteers [203]) might benefit from SSRIs even though there is no evidence backing this hypothesis. However, any clinical research result will have this 'limitation'. Specific patients might benefit from any given intervention even though valid research results have shown that this intervention 'on average' is ineffective or even harmful. All trials were at high risk of bias per several bias risk domains and especially the risk of incomplete outcome data, selective outcome reporting, and insufficient blinding bias may bias our review results. Our GRADE assessments show that due to the high risks of bias the quality of the evidence must be regarded as very low. The high risks of bias question the validity of our meta-analysis results as high risk of bias trials tend to overestimate benefits and underestimate harms [194, 205]. The ‘true’ effect of SSRIs might not even be statistically significant.
We chose to include all SSRIs in our primary analysis. We did this to increase the statistical power and precision and to be able to compare the effects of the different SSRIs in subgroup analysis. Comparing the different SSRIs in test for subgroup differences did not show significant differences, indicating the effects (or lack of effects) of the different SSRIs are similar. Nevertheless, we cannot rule out that certain SSRIs may have beneficial or harmful effects that we have not identified in this review due to lack of relevant data. We identified very limited data on the effects of SSRIs on long-term outcomes, suicidal behaviour, and quality of life, so the effects of SSRIs on these outcomes are unclear. E.g., we only identified six trials assessing quality of life which substantially increase the risk of selective outcome reporting bias and thereby limit the validity of the meta-analysis result. Furthermore, the trialists did not use the same questionnaire. Quality of life is without question an outcome with great relevance to the patient and we urge future trialists to assess quality of life. However, any given quality of life questionnaire must be validated (shown to be correlated to, e.g., suicidal behaviour or other clinical events) before valid conclusions may be drawn based on this outcome. It must be shown that scores on a given questionnaire do reflect the actual ‘quality of life’. Valid consensus on choosing the optimal quality of life assessment method does not exist and this is a limitation of assessing quality of life in depressed patients. Our eight-step procedure used to assess if the thresholds for statistical and clinical significance are crossed, is based on generally accepted and validated methodology but the use of the eight-step procedure has not yet been validated in simulation studies or empirical studies [12, 13]. Even though the eight-step procedure has been used in several systematic reviews it is not universally accepted. This may be a limitation of our methodology.
The Committee for Medicinal Products for Human Use (CHMP) concluded”……… that, as no public health concerns have been identified, no regulatory action is necessary on the basis of Kirsch et al.'s findings” when the latter team questioned the benefits of antidepressants [182]. Per our results, we now believe that there is valid evidence for a public concern regarding the effects of SSRIs. We agree with Andrews et al. that that antidepressants seem to do more harm than good [206]. We have clearly shown that SSRIs significantly increase the risks of both serious and several non-serious adverse events. The observed harmful effects seem to outweigh the potential small beneficial clinical effects of SSRIs, if they exist. Our results confirm the findings from other studies questioning the effects of SSRIs [8, 207], but are in contrast to the results of other reviews concluding that SSRIs are effective interventions for depression [3, 6, 10, 208]. However, our present analyses represent the most comprehensive systematic review on the topic and we hope it may guide clinical practice.
Conclusions
SSRIs versus placebo seem to have statistically significant effects on depressive symptoms, but the clinical significance of these effects seems questionable and all trials were at high risk of bias. Furthermore, SSRIs versus placebo significantly increase the risk of both serious and non-serious adverse events. Our results show that the harmful effects of SSRIs versus placebo for major depressive disorder seem to outweigh any potentially small beneficial effects.
References
Gualano MR, Bert F, Mannocci A, La Torre G, Zeppegno P, Siliquini R. Consumption of antidepressants in Italy: recent trends and their significance for public health. Psychiatr Serv. 2014;65(10):1226–31.
Wise J. GPs in England prescribed 2.7 million extra antidepressants during 2012 recession. BMJ. 2014;348:g3607.
Gibbons RD, Hur K, Brown CH, Davis JM, Mann JJ. Benefits from antidepressants: synthesis of 6-week patient-level outcomes from double-blind placebo-controlled randomized trials of fluoxetine and venlafaxine. Arch Gen Psychiatry. 2012;69(6):572–9.
Fournier JC, DeRubeis RJ, Hollon SD, Dimidjian S, Amsterdam JD, Shelton RC, Fawcett J. Antidepressant drug effects and depression severity: a patient-level meta-analysis. JAMA. 2010;303(1):47–53.
Khan A, Leventhal RM, Khan SR, Brown WA. Severity of depression and response to antidepressants and placebo: an analysis of the Food and Drug Administration database. J Clin Psychopharmacol. 2002;22(1):40–5.
Undurraga J, Baldessarini RJ. Randomized, placebo-controlled trials of antidepressants for acute major depression: thirty-year meta-analytic review. Neuropsychopharmacol. 2012;37(4):851–64.
Turner EH, Matthews AM, Linardatos E, Tell RA, Rosenthal R. Selective publication of antidepressant trials and its influence on apparent efficacy. N Engl J Med. 2008;358(3):252–60.
Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT. Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS Med. 2008;5(2):e45.
Arroll B, Elley CR, Fishman T, Goodyear-Smith FA, Kenealy T, Blashki G, Kerse N, MacGillivray S. Antidepressants versus placebo for depression in primary care. Cochrane Database of Syst Rev. 2009.
Wilson K, Mottram PG, Sivananthan A, Nightingale A. Antidepressants versus placebo for the depressed elderly. Cochrane Database Syst Rev. 2001.
Englev E, Petersen KP. ICH-GCP Guideline: quality assurance of clinical trials. Status and perspectives. Dan Med J. 2003;165(16):1659–62.
Jakobsen JC, Gluud C, Winkel P, Lange T, Wetterslev J. The thresholds for statistical and clinical significance - a five-step procedure for evaluation of intervention effects in randomised clinical trials. BMC Med Res Methodol. 2014;14:34.
Jakobsen JC, Wetterslev J, Winkel P, Lange T, Gluud C. Thresholds for statistical and clinical significance in systematic reviews with meta-analytic methods. BMC Med Res Methodol. 2014;14(1):120.
Jakobsen JC, Lindschou J, Hellmuth S, Schou A, Krogh J, Gluud C. The effects of selective serotonin reuptake inhibitors versus no intervention, placebo, or ‘active’ placebo in patients with major depressive disorder. A systematic review of randomised clinical trials with meta-analyses and trial sequential analyses. PROSPERO 2013:CRD42013004420 Available from http://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42013004420.
Moncrieff J, Wessely S, Hardy R. Active placebos versus antidepressants for depression. Coch Database Syst Rev. 2004;1:CD003012.
Higgins JPT, Green S. The Cochrane Handbook for Systematic Reviews of Interventions, Version 5.1.0. The Cochrane Collaboration 2011, Available from http://www.cochrane-handbook.org/.
Jakobsen JC, Lindschou J, Hellmuth S, Schou A, Krogh J, Gluud C. The effects of selective serotonin reuptake inhibitors versus no intervention, placebo, or ‘active’ placebo in patients with major depressive disorder. A systematic review of randomised clinical trials with meta-analyses and trial sequential analyses. PROSPERO 2013:CRD42013004420 Available from http://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42013004420 2013.
Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23(3):56–61.
Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–9.
Beck A, Steer RA, Brown GK. Beck depression inventory—second edition: manual. San Antonio: Psychological Corporation. 1996;4:561–71.
Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, Norris S, Falck-Ytter Y, Glasziou P, DeBeer H, et al. GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. J Clin Epidemiol. 2011;64(4):383–94.
DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7(3):177–88.
The Nordic Cochrane Centre (The Cochrane Collaboration): Review Manager (RevMan) [Computer program]. Version 5.3. In.; 2008.
Brok J, Thorlund K, Gluud C, Wetterslev J. Trial sequential analysis reveals insufficient information size and potentially false positive results in many meta-analysis. J Clin Epidemiol. 2008;61:763–9.
Wetterslev J, Thorlund K, Brok J, Gluud C. Trial sequential analysis may establish when firm evidence is reached in cumulative meta-analysis. J Clin Epidemiol. 2008;61(1):64–75.
Thorlund K, Devereaux PJ, Wetterslev J, Guyatt G, Ioannidis JP, Thabane L, Gluud LL, Als-Nielsen B, Gluud C. Can trial sequential monitoring boundaries reduce spurious inferences from meta-analyses? Int J Epidemiol. 2009;38(1):276–86.
Thorlund K, Imberger G, Walsh M, Chu R, Gluud C, Wetterslev J, Guyatt G, Devereaux PJ, Thabane L. The number of patients and events required to limit the risk of overestimation of intervention effects in meta-analysis – a simulation study. PLoS One. 2011;6:e25491.
Thorlund K, Engstrøm J, Wetterslev J, Brok J, Imberger G, Gluud C. User manual for trial sequential analysis (TSA). Copenhagen: Copenhagen Trial Unit, Centre for Clinical Intervention Research; 2011. p. 1–115. Available from http://www.ctu.dk/tsa.
Wetterslev J, Thorlund K, Brok J, Gluud C. Estimating required information size by quantifying diversity in random-effects model meta-analyses. BMC Med Res Methodol. 2009;9:86.
Higgins J. Convincing evidence from controlled and uncontrolled studies on the lipid-lowering effect of a statin [editorial]. Cochrane Database Syst Rev. 2012;12:ED000049.
National Institute for Clinical Excellence. Depression: Mangement of Depression in Primary and Secundary Care. London: National Institute for Clinical Excellence; 2004. p. 640.
Byerley WF, Reimherr FW, Wood DR, Grosser BI. Fluoxetine, a selective serotonin uptake inhibitor, for the treatment of outpatients with major depression. J Clin Psychopharmacol. 1988;8:112–5.
Davidson JRT, Gadde KM, Fairbank JA, Ranga Rama KK, Califf RM, Binanay C, Parker CB, Pugh N, Hartwell TD, Vitiello B, et al. Effect of Hypericum perforatum (St John's wort) in major depressive disorder: A randomized controlled trial. JAMA. 2002;287(14):10.
Fava M, Alpert J, Nierenberg AA, Mischoulon D, Otto MW, Zajecka J, Murck H, Rosenbaum JF. A Double-blind, randomized trial of St John's wort, fluoxetine, and placebo in major depressive disorder. J Clin Psychopharmacol. 2005;25:441–7.
Fava M, Amsterdam JD, Deltito JA, Salzman C, Schwaller M, Dunner DL. A double-blind study of paroxetine, fluoxetine, and placebo in outpatients with major depression. Ann Clin Psychiatry. 1998;10:145–50.
Feighner JP, Boyer WF, Merideth CH, Hendrickson GG. A double-blind comparison of fluoxetine, imipramine and placebo in outpatients with major depression. Int Clin Psychopharmacol. 1989;4:127–34.
Godlewska BR, Norbury R, Selvaraj S, Cowen PJ, Harmer CJ. Short-term SSRI treatment normalises amygdala hyperactivity in depressed patients. Psychol Med. 2012;42:2609–17.
Hunter AM, Cook IA, Greenwald SD, Tran ML, Miyamoto KN, Leuchter AF. The antidepressant treatment response index and treatment outcomes in a placebo-controlled trial of fluoxetine. J Clin Neurophysiol. 2011;28:478–82.
Itil TM, Shrivastava RK, Mukherjee S. A double-blind placebo-controlled study of fluvoxamine and imipramine in out-patients with primary depression. Brit J Clin Pharmacol. 1983;15(Suppl#3):1983.
Jindal RD, Friedman ES, Berman SR, Fasiczka AL, Howland RH, Thase ME. Effects of sertraline on sleep architecture in patients with depression. J Clin Psychopharmacol. 2003;23:540–8.
Katz MM, Tekell JL, Bowden CL, Brannan S, Houston JP, Berman N, Frazer A. Onset and early behavioral effects of pharmacologically different antidepressants and placebo in depression. Neuropsychopharmacol. 2004;29:566–79.
McGrath PJ, Stewart JW, Janal MN, Petkova E, Quitkin FM, Klein DF. A placebo-controlled study of fluoxetine versus imipramine in the acute treatment of atypical depression. Am J Psychiatry. 2000;157:344–50.
Muijen M, Roy D, Silverstone T, Mehmet A, Christie M. A comparative clinical trial of fluoxetine, mianserin and placebo in depressed outpatients. Acta Psychiatr Scand. 1988;78:384–90.
Petrakis I, Carroll KM, Nich C, Gordon L, Kosten T, Rounsaville B. Fluoxetine treatment of depressive disorders in methadone-maintained opioid addicts. Drug Alcohol Depend. 1998;50:221–6.
Rapaport MH, Schneider LS, Dunner DL, Davies JT, Pitts CD. Efficacy of controlled-release paroxetine in the treatment of late-life depression. J Clin Psychiatry. 2003;64:1065–74.
Roth D, Mattes J, Sheehan KH, Sheehan DV. A double-blind comparison of fluvoxamine, desipramine and placebo in outpatients with depression. Prog Neuro Psychopharmacol Biol Psychiat. 1990;14:929–39.
Roy A. Placebo-controlled study of sertraline in depressed recently abstinent alcoholics. Biol Psychiatry. 1998;44:633–7.
Schneider LS, Nelson JC, Clary CM, Newhouse P, Krishnan KR, Shiovitz T, Weihs K, Sertraline Elderly Depression Study G. An 8-week multicenter, parallel-group, double-blind, placebo-controlled study of sertraline in elderly outpatients with major depression. Am J Psychiatry. 2003;160:1277–85.
Sheehan DV, Nemeroff CB, Thase ME, Entsuah R: Placebo-controlled inpatient comparison of venlafaxine and fluoxetine for the treatment of major depression with melancholic features. Int Clin Psychopharmacol 2009, 24(2):March.
Mischoulon D, Price LH, Carpenter LL, Tyrka AR, Papakostas GI, Baer L, Dording CM, Clain AJ, Durham K, Walker R, et al. A double-blind, randomized, placebo-controlled clinical trial of S-adenosyl-L-methionine (SAMe) versus escitalopram in major depressive disorder. J Clin Psychiatry. 2014;75(4):370–6.
Barber JP, Barrett MS, Gallop R, Rynn MA, Rickels K. Short-term dynamic psychotherapy versus pharmacotherapy for major depressive disorder: a randomized, placebo-controlled trial. J Clin Psychol. 2012;73(1):66–73.
Binneman B, Feltner D, Kolluri S, Shi Y, Qiu R, Stiger T. A 6-week randomized, placebo-controlled trial of CP-316,311 (a selective CRH1 antagonist) in the treatment of major depression. Am J Psychiatry. 2008;165:617–20.
Blumenthal JA, Babyak MA, Doraiswamy PM, Watkins L, Hoffman BM, Barbour KA, Herman S, Craighead WE, Brosse AL, Waugh R, et al. Exercise and pharmacotherapy in the treatment of major depressive disorder. Psychosomatic Med. 2007;69:587–96.
Bose A, Li D, Gandhi C. Escitalopram in the acute treatment of depressed patients aged 60 years or older. Am J Geriatr Psychiatry. 2008;16:14–20.
Detke MJ, Wiltse CG, Mallinckrodt CH, McNamara RK, Demitrack MA, Bitter I. Duloxetine in the acute and long-term treatment of major depressive disorder: a placebo- and paroxetine-controlled trial. Eur Neuropsychopharmacol. 2004;14(6):457–70.
Dube S, Dellva MA, Jones M, Kielbasa W, Padich R, Saha A, Rao P: A study of the effects of LY2216684, a selective norepinephrine reuptake inhibitor, in the treatment of major depression. J Psychiatric Res 2010, 44(6):April.
Edwards JG, Goldie A. Placebo-controlled trial of paroxetine in depressive illness. Hum Psychopharmacol. 1993;8(3):1993.
Fabre LF. A 6-week, double-blind trial of paroxetine, imipramine, and placebo in depressed outpatients. J Clin Psychiatry. 1992;53(Suppl):40–3.
Fabre LF, Abuzzahab FS, Amin M, Claghorn JL, Mendels J, Petrie WM, Dub S, Small JG. Sertraline safety and efficacy in major depression: A double-blind fixed-dose comparison with placebo. Biol Psychiatry. 1995;38(9):1995.
Higuchi T, Hong JP, Jung HY, Watanabe Y, Kunitomi T, Kamijima K. Paroxetine controlled-release formulation in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled study in Japan and Korea. Psychiatry Clin Neurosci. 2011;65(7):655–63.
Kranzler HR, Mueller T, Cornelius J, Pettinati HM, Moak D, Martin PR, Anthenelli R, Brower KJ, O'Malley S, Mason BJ, et al. Sertraline treatment of co-occurring alcohol dependence and major depression. J Clin Psychopharmacol. 2006;26:13–20.
Lydiard RB, Stahl SM, Hertzman M, Harrison WM. A double-blind, placebo-controlled study comparing the effects of sertraline versus amitriptyline in the treatment of major depression. J Clin Psychiatry. 1997;58:484–91.
Rapaport MH, Lydiard RB, Pitts CD, Schaefer D, Bartolic EI, Iyengar M, Carfagno M, Lipschitz A. Low doses of controlled-release paroxetine in the treatment of late-life depression: a randomized, placebo-controlled trial. J Clin Psychiatry. 2009;70:46–57.
Reimherr FW, Chouinard G, Cohn CK, Cole JO, Itil TM, Lapierre YD, Masco HL, Mendels J. Antidepressant efficacy of sertraline: a double-blind, placebo- and amitriptyline-controlled, multicenter comparison study in outpatients with major depression. J Clin Psychiatry. 1990;51(Suppl B):18–27.
Sramek JJ, Kashkin K, Jasinsky O, Kardatzke D, Kennedy S, Cutler NR. Placebo-controlled study of ABT-200 versus fluoxetine in the treatment of major depressive disorder. Depress. 1995;3(4):1995.
Burke WJ, Gergel I, Bose A. Fixed-dose trial of the single isomer SSRI escitalopram in depressed outpatients. J Clin Psychiatry. 2002;63:331–6.
Cassano GB, Conti L, Massimetti G, Mengali F, Waekelin JS, Levine J. Use of a standardized documentation system (BLIPS/BDP) in the conduct of a multicenter international trial comparing fluvoxamine, imipramine, and placebo. Psychopharmacol Bull. 1986;22:52–8.
Cohn JB, Wilcox C. A comparison of fluoxetine, imipramine, and placebo in patients with major depressive disorder. J Clin Psychiatry. 1985;46:26–31.
Coleman CC, King BR, Bolden WC, Book MJ, Segraves RT, Richard N, Ascher J, Batey S, Jamerson B, Metz A. A placebo-controlled comparison of the effects on sexual functioning of bupropion sustained release and fluoxetine. Clin Therapeutics. 2001;23:1040–58.
Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress Anxiety. 2000;11:58–65.
DeRubeis RJ, Hollon SD, Amsterdam JD, Shelton RC, Young PR, Salomon RM, O'Reardon JP, Lovett ML, Gladis MMBL, Gallop R. Cognitive therapy vs medications in the treatment of moderate to severe depression. Arch Gen Psychiatry. 2005;62:409–16.
Dominguez RA, Goldstein BJ, Jacobson AF, Steinbook RM. A double-blind placebo-controlled study of fluvoxamine and imipramine in depression. J Clin Psychiatry. 1985;46:84–7.
Fabre LF, Crismon L. Efficacy of fluoxetine in outpatients with major depression. Curr Ther Res Clin Exp. 1985;37(1):1985.
Lydiard RB, Laird LK, Morton Jr, Steele TE, Kellner C, Laraia MT, Ballenger JC. Fluvoxamine, imipramine, and placebo in the treatment of depressed outpatients: Effects on depression. Psychopharmacol Bull. 1989;25(1):68.
March JS, Kobak KA, Jefferson JW, Mazza J, Greist JH. A double-blind, placebo-controlled trial of fluvoxamine versus imipramine in outpatients with major depression. J Clin Psychiatry. 1990;51:200–2.
Mayberg HS, Silva JA, Brannan SK, Tekell JL, Mahurin RK, McGinnis S, Jerabek PA. The functional neuroanatomy of the placebo effect. Am J Psychiatry. 2002;159:728–37.
Miller SM, Naylor GJ, Murtagh M, Winslow G. A double-blind comparison of paroxetine and placebo in the treatment of depressed patients in a psychiatric outpatient clinic. Acta Psychiatr Scand. 1989;350:143–4.
Nemeroff CB, Thase ME: A double-blind, placebo-controlled comparison of venlafaxine and fluoxetine treatment in depressed outpatients. J Psychiatr Res 2007, 41(3–4):April/June.
Norton KR, Sireling LI, Bhat AV, Rao B, Paykel ES: A double-blind comparison of fluvoxamine, imipramine and placebo in depressed patients. J Affect Disord 1984, 7(3–4):Dec.
Rickels K, Amsterdam JD, Avallone MF. Fluoxetine in major depression: A controlled study. Curr Ther Res Clin Exp. 1986;39(4):1986.
Rudolph RL, Feiger AD. A double-blind, randomized, placebo-controlled trial of once-daily venlafaxine extended release (XR) and fluoxetine for the treatment of depression. J Affect Disord. 1999;56:171–81.
Schatzberg A, Roose S. A double-blind, placebo-controlled study of venlafaxine and fluoxetine in geriatric outpatients with major depression. Am J Geriatr Psychiatry. 2006;14:361–70.
Shrivastava RK, Shrivastava SH, Overweg N, Blumhardt CL. A double-blind comparison of paroxetine, imipramine, and placebo in major depression. J Clin Psychiatry. 1992;53(Suppl):48–51.
Smith WT, Glaudin V. A placebo-controlled trial of paroxetine in the treatment of major depression. J Clin Psychiatry. 1992;53(Suppl):36–9.
Tyrer P, Marsden CA, Casey P, Seivewright N. Clinical efficacy of paroxetine in resistant depression. J Psychopharmacol. 1987;1(4):1987.
Fabre LF, Putman HP. A fixed-dose clinical trial of fluoxetine in outpatients with major depression. J Clin Psychiatry. 1987;48:406–8.
Feighner JP, Boyer WF. Paroxetine in the treatment of depression: a comparison with imipramine and placebo. Acta Psychiatr Scand. 1989;350:125–9.
Feighner JP, Boyer WF, Meredith CH, Hendrickson GG. A placebo-controlled inpatient comparison of fluvoxamine maleate and imipramine in major depression. Int Clin Psychopharmacol. 1989;4:239–44.
Claghorn JL. The safety and efficacy of paroxetine compared with placebo in a double-blind trial of depressed outpatients. J Clin Psychiatry. 1992;53(Suppl):33–5.
Claghorn JL, Earl CQ, Walczak DD, Stoner KA, Wong LF, Kanter D, Houser VP. Fluvoxamine maleate in the treatment of depression: a single-center, double-blind, placebo-controlled comparison with imipramine in outpatients. J Clin Psychopharmacol. 1996;16:113–20.
Cohn JB, Crowder JE, Wilcox CS, Ryan PJ. A placebo- and imipramine-controlled study of paroxetine. Psychopharmacol Bull. 1990;26:185–9.
Dunbar GC, Cohn JB, Fabre LF, Feighner JP, Fieve RR, Mendels J, Shrivastava RK. A comparison of paroxetine, imipramine and placebo in depressed out-patients. Brit J Psychiatry. 1991;159:394–8.
Fabre L, Birkhimer LJ, Zaborny BA, Wong LF, Kapik BM. Fluvoxamine versus imipramine and placebo: a double-blind comparison in depressed patients. Int Clin Psychopharmacol. 1996;11:119–27.
Feighner JP, Overõ K. Multicenter, placebo-controlled, fixed-dose study of citalopram in moderate-to-severe depression. J Clin Psychiatry. 1999;60:824–30.
Griebel G, Beeské S, Stahl SM. The vasopressin V(1b) receptor antagonist SSR149415 in the treatment of major depressive and generalized anxiety disorders: results from 4 randomized, double-blind, placebo-controlled studies. J Clin Psychiatry. 2012;73:1403–11.
Kiev A. A double-blind, placebo-controlled study of paroxetine in depressed outpatients. J Clin Psychiatry. 1992;53(Suppl):27–9.
Olie JP, Gunn KP, Katz E. A double-blind placebo-controlled multicentre study of sertraline in the acute and continuation treatment of major depression. Eu Psychiatry. 1997;12(1):1997.
Peselow ED, Stanley M, Filippi AM, Barouche F, Goodnick P, Fieve RR. The predictive value of the dexamethasone suppression test. A placebo-controlled study. Brit J Psychiatry. 1989;155:667–72.
Rickels K, Amsterdam J, Clary C, Fox I, Schweizer E, Weise C. The efficacy and safety of paroxetine compared with placebo in outpatients with major depression. J Clin Psychiatry. 1992;53(Suppl):30–2.
Stahl SM. Placebo-controlled comparison of the selective serotonin reuptake inhibitors citalopram and sertraline. Biol Psychiatry. 2000;48:894–901.
Trivedi MH, Pigotti TA, Perera P, Dillingham KE, Carfagno ML, Pitts CD. Effectiveness of low doses of paroxetine controlled release in the treatment of major depressive disorder. J Clin Psychiatry. 2004;65:1356–64.
Forest Laboratories Inc: SCT-MD-01 (Unpublished Data) Forest Laboratories Inc. Study report from U.S. Food and Drug Administration. 2001.
Hoffman BM, Blumenthal JA, Babyak MA, Smith PJ, Rogers SD, Doraiswamy PM, Sherwood A. Exercise fails to improve neurocognition in depressed middle-aged and older adults. Med Sci Sports Exerc. 2008;40:1344–52.
Brunoni AR, Valiengo L, Baccaro A, Zanão TA, de Oliveira JF, Goulart A, Boggio PS, Lotufo PA, Benseñor IM, Fregni F. The sertraline vs. electrical current therapy for treating depression clinical study: results from a factorial, randomized, controlled trial. JAMA Psychiat. 2013;70:383–91.
Kasper S, Ebert B, Larsen K, Tonnoir B. Combining escitalopram with gaboxadol provides no additional benefit in the treatment of patients with severe major depressive disorder. European Neuropsychopharmacol (conference Publication). 2011;21:S408.
Wade A, Michael LO, Bang HK. Escitalopram 10 mg/day is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol. 2002;17:95–102.
Silverstone PH, Ravindran A. Once-daily venlafaxine extended release (XR) compared with fluoxetine in outpatients with depression and anxiety. Venlafaxine XR 360 Study Group. J Clin Psychiatry. 1999;60:22–8.
Andreoli V, Caillard V, Deo RS, Rybakowski JK, Versiani M. Reboxetine, a new noradrenaline selective antidepressant, is at least as effective as fluoxetine in the treatment of depression. J Clin Psychopharmacol. 2002;22:393–9.
O'Flynn K, O'Keane V, Lucey JV, Dinan TG. Effect of fluoxetine on noradrenergic mediated growth hormone release: a double blind, placebo-controlled study. Biol Psychiatry. 1991;30:377–82.
Reynolds CF, Smith GS, Dew MA, Mulsant BH, Miller MD, Schlernitzauer M, Stack JA, Houck PR, Pollock BG. Accelerating symptom-reduction in late-life depression: a double-blind, randomized, placebo-controlled trial of sleep deprivation. Am J Geriatr Psychiatry. 2005;13:353–8.
Roose SP, Sackeim HA, Krishnan KR, Pollock BG, Alexopoulos G, Lavretsky H, Katz IR, Hakkarainen H, Old Old Depression Study G. Antidepressant pharmacotherapy in the treatment of depression in the very old: a randomized, placebo-controlled trial. Am J Psychiatry. 2004;161:2050–9.
Wang G, McIntyre A, Eriksson H. A randomized, double-blind study of the efficacy and tolerability of extended-release quetiapine fumarate (quetiapine XR) monotherapy in patients with major depressive disorder. Neuropsychiatr Dis Treat. 2014, 10.
Kasper S, de Swart H, Friis AH. Escitalopram in the treatment of depressed elderly patients. Am J Geriatr Psychiatry. 2005;13:884–91.
Learned S, Graff O, Roychowdhury S, Moate R, Krishnan KR, Archer G, Modell JG, Alexander R, Zamuner S, Lavergne A, et al. Efficacy, safety, and tolerability of a triple reuptake inhibitor GSK372475 in the treatment of patients with major depressive disorder: two randomized, placebo- and active-controlled clinical trials. J Psychopharmacol. 2012;26:653–62.
Mancino MJ, McGaugh J, Chopra MP, Guise JB, Cargile C, Williams DK, Thostenson J, Kosten TR, Sanders N, Oliveto A. Clinical efficacy of sertraline alone and augmented with gabapentin in recently abstinent cocaine-dependent patients with depressive symptoms. J Clin Psychopharmacol. 2014;34:234–9.
Pettinati HM, Oslin DW, Kampman KM, Dundon WD, Xie H, Gallis TL, Dackis CA, O'Brien CP. A double-blind, placebo-controlled trial combining sertraline and naltrexone for treating co-occurring depression and alcohol dependence. Am J Psychiatry. 2010;167:668–75.
Ratti E, Bellew K, Bettica P, Bryson H, Zamuner S, Archer G, Squassante L, Bye A, Trist D, Krishnan KR, et al. Results from 2 randomized, double-blind, placebo-controlled studies of the novel NK1 receptor antagonist casopitant in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31:727–33.
Ravindran AV, Teehan MD, Bakish D, Yatham L, Oreilly R, Fernando ML, Manchanda R, Charbonneau Y, Buttars J. The impact of sertraline, desipramine, and placebo on psychomotor functioning in depression. Human Psychopharmacol. 1995;10(4):273–81.
315 (Unpublished Data). Study report from U.S. Food and Drug Administration. In.; 1991.
Adamson SJ, Sellman JD, Foulds J, Frampton CM, Deering D, Dunn A, Berks J, Nixon L, Cape G. A randomized trial of combined citalopram and naltrexone for nonabstinent outpatients with co-occurring alcohol dependence and major depression. J Clin Psychopharmacol. 2015;35:143–9.
Ball WA, Snavely DB, Hargreaves RJ, Szegedi A, Lines C, Reines SA. Addition of an Nk1 receptor antagonist to an Ssri did not enhance the antidepressant effects of Ssri monotherapy: results from a randomized clinical trial in patients with major depressive disorder. Hum Psychopharmacol. 2014;29:568–77.
Chen YM, Huang XM, Thompson R, Zhao YB. Clinical features and efficacy of escitalopram treatment for geriatric depression. J Int Med Res. 2011;39(5):2011.
Clayton AH, Croft H, Horrigan JP, Wightman DS, Krishen A, Richard NE, Modell JG. Bupropion extended release compared with escitalopram: Effects on sexual functioning and antidepressant efficacy in 2 randomized, double-blind, placebo-controlled studies. J Clin Psychiatry. 2006;67:736–46.
Coleman CC, Cunningham LA, Foster VJ, Batey SR, Donahue RM, Houser TL, Ascher JA. Sexual dysfunction associated with the treatment of depression: a placebo-controlled comparison of bupropion sustained release and sertraline treatment. Ann Clin Psychiatry. 1999;11:205–15.
Croft H, Settle E, Houser T, Batey SR, Donahue RM, Ascher JA. A placebo-controlled comparison of the antidepressant efficacy and effects on sexual functioning of sustained-release bupropion and sertraline. Clin Ther. 1999;21:643–58.
Doogan DP, Langdon CJ. A double-blind, placebo-controlled comparison of sertraline and dothiepin in the treatment of major depression in general practice. Int Clin Psychopharmacol. 1994;9:95–100.
Fabre LF, Clayton AH, Smith LC, Goldstein I, Derogatis LR. The Effect of Gepirone-ER in the Treatment of Sexual Dysfunction in Depressed Men. J Sex Med. 2012, 9(3):March.
Golden RN, Nemeroff CB, McSorley P, Pitts CD, DubÇ EM. Efficacy and tolerability of controlled-release and immediate-release paroxetine in the treatment of depression. J Clin Psychiatry. 2002;63:577–84.
Hunter AM, Leuchter AF, Cook IA, Abrams M. Brain functional changes (QEEG cordance) and worsening suicidal ideation and mood symptoms during antidepressant treatment. Acta Psychiatr Scand. 2010;122:461–9.
Lepola UM, Loft H, Reines EH. Escitalopram (10–20 mg/day) is effective and well tolerated in a placebo-controlled study in depression in primary care. Int Clin Psychopharmacol. 2003;18:211–7.
Leuchter AF, Morgan M, Cook IA, Dunkin J, Abrams M, Witte E. Pretreatment neurophysiological and clinical characteristics of placebo responders in treatment trials for major depression. Psychopharmacology (Berl). 2004;177:15–22.
Chan A-W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža-Jerić K, Hróbjartsson A, Mann H, Dickersin K, Berlin JA, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–7.
Mao JJ, Xie SX, Zee J, Soeller I, Li QS, Rockwell K, Amsterdam JD. Rhodiola rosea versus sertraline for major depressive disorder: A randomized placebo-controlled trial. Phytomedicine. 2015;22:394–9.
Mathews M, Gommoll C, Nunez R, Khan A. Efficacy and safety of vilazodone 20 and 40 Mg in major depressive disorder: a randomized, double-blind, placebo-controlled trial. Int Clin Psychopharmacol. 2015;30:67–74.
Mendels J, Kiev A, Fabre LF. Double-blind comparison of citalopram and placebo in depressed outpatients with melancholia. Depress Anxiety. 1999;9:54–60.
Montgomery SA, Rasmussen JG, Lyby K, Connor P, Tanghõj P. Dose response relationship of citalopram 20 mg, citalopram 40 mg and placebo in the treatment of moderate and severe depression. Int Clin Psychopharmacol. 1992;6 Suppl 5:65–70.
MY-1045/BRL-029060/1 (PAR128) (Unpublished Data) GlaxoSmithKline. A Multicenter, Randomized, Double-Blind, Placebo-Controlled Comparison of Paroxetine and Fluoxetine in the Treatment of Major Depressive Disorder. http://www.gsk-clinicalstudyregister.com/files2/2821.pdf 2005, 1:1–5.
MY-1043/BRL-029060/115 (Unpublished Data) GlaxoSmithKline. A multicenter, randomized, double-blind, placebo-controlled comparison of paroxetine and fluoxetine in the treatment of major depressive disorder. http://www.gsk-clinicalstudyregister.com/files2/2060.pdf 2005, 175:1–5.
Rath A, Ngwabyt S, Durisic S, Garattini S, Neugebauer EAM, Laville M, Jakobsen JC, Kubiac C, DeMotes-Mainard J, Gluud C: Specific barriers to the conduct of randomised clinical trials within rare diseases - the Europeean Clinical Research Infrastructure (ECRIN) perspective. TRIALS, to be submitted 2015.
AstraZeneca. AZD7268 Safety and Tolerability Study. In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2015 Jun 19]. https://clinicaltrials.gov/ct2/show/results/NCT01020799?term=NCT01020799&rank=1. In.; 2012.
Nierenberg, Greist JH, Mallinckrodt CH, Prakash A, Sambunaris A, Tollefson GD, Wohlreich MM. Duloxetine versus escitalopram and placebo in the treatment of patients with major depressive disorder: onset of antidepressant action, a non-inferiority study. Curr Med Res Opinion. 2007;23:401–16.
PAR 01-001(Unpublished Data). Study report from U.S. Food and Drug Administration. In.; 1992.
PAR 09 (Unpublished Data). Study report from U.S. Food and Drug Administration. In.; 1992.
Nyth aL, Gottfries CG, Lyby K, Smedegaard-Andersen L, Gylding-Sabroe J, Kristensen M, Refsum HE, Ofsti E, Eriksson S, Syversen S. A controlled multicenter clinical study of citalopram and placebo in elderly depressed patients with and without concomitant dementia. Acta Psychiatr Scand. 1992;86:138–45.
PAR 29060.07.001 (Unpublished Data) GlaxoSmithKline. A Double-Blind Comparison of Paroxetine, Amitriptyline, and Placebo in Inpatients with Major Depressive Disorder with Melancholia. http://www.gsk-clinicalstudyregister.com/files2/1866.pdf 2008:1–4.
Perahia DGS, Wang F, Mallinckrodt CH, Walker DJ, Detke MJ. Duloxetine in the treatment of major depressive disorder: a placebo- and paroxetine-controlled trial. Eu Psychiatry. 2006;21:367–78.
Peselow ED, Lautin A, Wolkin A. The dexamethasone suppression test and response to placebo. J Clin Psychopharmacol. 1986;6(5):1986.
Porro V, Fiorenzioni S, Menga C, De CA, Bertolino A. Single-blind comparison of the efficacy of fluvoxamine versus placebo in patients with depressive syndrome. Curr Therapeutic Res. 1988;43(4):1988.
SCT-MD-26 (Unpublished Data) Forest Research Institute. Two-week double-blind placebo controlled study of escitalopram in the treatment of severe major depression. 2005, 135:2–4.
SCT-MD-27 (Unpublished Data) Forest Research Institute. A double-blind flexible dose comparison of escitalopram sertraline and placebo in the treatment of major depressive disorder. 2005, 91:5–7.
SCT-MD-35 (Unpublished Data) Forest Research Institute. Fixed-dose comparison of escitalopram combination in adult patients with major depressive disorder. 2007:1–28.
Walczak DD, Apter JT, Halikas JA, Borison RL, Carman JS, Post GL, Patrick R, Cohn JB, Cunningham LA, Rittberg B, et al. The oral dose-effect relationship for fluvoxamine: a fixed-dose comparison against placebo in depressed outpatients. Ann Clin Psychiatry. 1996;8:139–51.
Zitman FG, CouvÇe JE. Chronic benzodiazepine use in general practice patients with depression: an evaluation of controlled treatment and taper-off: report on behalf of the Dutch Chronic Benzodiazepine Working Group. Brit J Psychiatry. 2001;178:317–24.
Hutton B, Salanti G, Caldwell DM, Chaimani A, Schmid CH, Cameron C, Ioannidis JP, Straus S, Thorlund K, Jansen JP, et al. The PRISMA extension statement for reporting of systematic reviews incorporating network meta-analyses of health care interventions: checklist and explanations. Ann Intern Med. 2015;162(11):777–84.
29060/785 (Unpublished Data) GlaxoSmithKline. A double-blind, placebo-controlled, fixed-dosage study comparing the efficacy and tolerability of paroxetine CR and citalopram to placebo in the treatment of Major Depressive Disorder with anxiety. http://www.gsk-clinicalstudyregister.com/files2/2202.pdf 2005:1–5.
Cornelius JR, Salloum IM, Ehler JG, Jarrett PJ, Cornelius MD, Perel JM, Thase ME, Black A. Fluoxetine in depressed alcoholics. A double-blind, placebo-controlled trial. Arch Gen Psychiatry. 1997;54:700–5.
Goldstein DJ, Lu Y, Detke MJ, Wiltse C, Mallinckrodt C, Demitrack M. Duloxetine in the treatment of depression. J Clin Psychopharmacol. 2004;24:389–99.
Goldstein DJ, Ph D, Mallinckrodt C, Ph D, Lu Y, Ph D, Demitrack MA. Duloxetine in the treatment of major depressive disorder: a double-blind clinical trial. J Clin Psychiatry. 2002;63(3):225–31.
Kaddurah-Daouk R, Boyle SH, Matson W, Sharma S, Matson S, Zhu H, Bogdanov MB, Churchill E, Krishnan RR, Rush aJ, et al. Pretreatment metabotype as a predictor of response to sertraline or placebo in depressed outpatients: a proof of concept. Translat Psychiatry. 2011;1:e26.
Kennedy S. Flibanserin: initial evidence of efficacy on sexual dysfunction, in patients with major depressive disorder. J Sex Med. 2010;7(10):3449–59.
Dubini A, Bosc M, Polin V. Noradrenaline-selective versus serotonin-selective antidepressant therapy: differential effects on social functioning. J Psychopharmacol. 1997;11:S17–23.
Tollefson GD, Bosomworth JC, Heiligenstein JH, Potvin JH. A double-blind, placebo-controlled clinical trial of fluoxetine in geriatric patients with major depression. Int Psychogeriatr. 1995;1:89–104.
Dunlop SR, Dornseif BE, Wernicke JF, Potvin JH. Pattern analysis shows beneficial effect of fluoxetine treatment in mild depression. Psychopharmacol Bull. 1990;26(2):1.
Schmitz JM, Averill P, Stotts AL, Moeller FG, Rhoades HM, Grabowski J. Fluoxetine treatment of cocaine-dependent patients with major depressive disorder. Drug Alcohol Depend. 2001;63:207–14.
89306 (Unpublished Data). Study report from U.S. Food and Drug Administration. In.; 1998.
Wakelin JS. Fluvoxamine in the treatment of the older depressed patient; double-blind, placebo-controlled data. Int Clin Psychopharmacol. 1986;1:221–30.
29060/448 (Unpublished Data) GlaxoSmithKline. A double-blind, placebo controlled trial to evaluate the clinical effects of immediate release paroxetine and modified release paroxetine in the treatment of major depression. URL: https://www.gsk-clinicalstudyregister.com/files2/2021.pdf. 2005.
NKF100096 (Unpublished Data) GlaxoSmithKline. A randomised, double-blind, double-dummy, parallel-group, placebo-controlled, forced dose titration study evaluating the efficacy and safety of a new chemical entity (NCE) and paroxetine in subjects with major depressive disorder (MDD). 2007:4–7.
89303 (Unpublished Data) Forest Laboratories Inc. Study report from U.S. Food and Drug Administration. In.; 1998.
Cohn JB, Wilcox CS. Paroxetine in major depression: a double-blind trial with imipramine and placebo. J Clin Psychiatry. 1992;53(Suppl):52–6.
Vittinghoff E, McCulloch CE. Relaxing the rule of ten events per variable in logistic and cox regression. Am J Epidemiol. 2007;165(6):710–8.
Tollefson GD, Holman SL. Analysis of the Hamilton Depression Rating Scale factors from a double-blind, placebo-controlled trial of fluoxetine in geriatric major depression. Int Clin Psychopharmacol. 1993;8:253–9.
CIT-MD-03 (Unpublished Data) Forest Laboratories Inc. A randomized double-blind placebo-controlled trial of citalopram in depressed patients at least 75 years of age. 2005:1–4.
Bech AT. An inventory for measuring depression. Arch Gen Psychiatry. 1961;4:561–71.
SND103288 (Unpublished Data) GlaxoSmithKline. A ten-week, multicentre, randomised, double-blind, placebo-and active-controlled, parallel-group, flexible-dose study evaluating the efficacy, safety and tolerability of GSK372475, a new chemical entity (NCE), or paroxetine compared to placebo in adult subjects diagnosed with major depressive disorder. URL: http://www.gsk-clinicalstudyregister.com/files2/21101.pdf. 2008:1–9.
WELL AK130926 (Unpublished Data) GlaxoSmithKline. A multicenter, double-blind, randomized, placebo-controlled comparison of the effects on sexual functioning of extended-release bupropion hydrochloride (300–450 mg) and escitalopram (10–20 mg) in outpatients with moderate to severe major depression over an eight-week treatment period. URL: http://www.gsk-clinicalstudyregister.com/files2/20466.pdf. 2007.
Sweeting MJ, Sutton AJ, Lambert PC. What to add to nothing? Use and avoidance of continuity corrections in meta-analysis of sparse data. Stat Med. 2004;23(9):1351–75.
Forest Laboratories Inc: SCT-MD-01. Unpublished 2001, Study report from FDA.
29060/810 (Unpublished Data) GlaxoSmithKline. A randomized, double-blind, parallel-group, placebo-controlled study evaluating efficacy and safety of paroxetine controlled release (12.5 and 25 mg/day) versus placebo in patients with major depressive disorder. URL: https://www.gsk-clinicalstudyregister.com/files2/2204.pdf. 2005.
Forest Laboratories. Safety and efficacy of vilazodone in major depressive disorder (VLZ-MD-01). In: ClinicalTrials.gov [Internet]. Bethesda (MD): National Library of Medicine (US). 2000- [cited 2015 Jun 19]. Available from: http://clinicaltrials.gov/show/NCT01473381 URL: https://clinicaltrials.gov/ct2/show/results/NCT01473381?term=NCT01473381&rank=1. In.; 2014.
SCT-MD-02 (Unpublished Data) Forest Laboratories Inc. Study report from U.S. Food and Drug Administration. In.; 2002.
Broich K. Committee for Medicinal Products for Human Use (CHMP) assessment on efficacy of antidepressants. Eur Neuropsychopharmacol. 2009;19(5):305–8.
Hunt SM, McKenna SP. The QLDS: a scale for the measurement of quality of life in depression. Health Policy. 1992;22(3):307–19.
Jakobsen JC, Gluud C. The necessity of randomized clinical trials. Br J Med Res. 2013;3(4):1453–68.
Rimer J, Dwan K, Lawlor Debbie A, Greig Carolyn A, McMurdo M, Morley W, Mead Gillian E. Exercise for depression. Coch Database Syst Rev. 2012;7:CD004366. pub004366.
Leucht S, Fennema H, Engel R, Kaspers-Janssen M, Lepping P, Szegedi A. What does the HAMD mean? J Affect Disord. 2013;148(2–3):243–8.
Moncrieff J, Kirsch I. Empirically derived criteria cast doubt on the clinical significance of antidepressant-placebo differences. Contemp Clin Trials. 2015;43:60–2.
Furukawa TA, Cipriani A, Atkinson LZ, Leucht S, Ogawa Y, Takeshima N, Hayasaka Y, Chaimani A, Salanti G. Placebo response rates in antidepressant trials: a systematic review of published and unpublished double-blind randomised controlled studies. Lancet Psychiatry. 2016;3(11):1059–66.
Hrobjartsson A, Gotzsche PC. Placebo interventions for all clinical conditions. Cochrane Database Syst Rev. 2010;1:CD003974.
Hrobjartsson A, Emanuelsson F, Skou Thomsen AS, Hilden J, Brorson S. Bias due to lack of patient blinding in clinical trials. A systematic review of trials randomizing patients to blind and nonblind sub-studies. Int J Epidemiol. 2014;43(4):1272–83.
Hróbjartsson A, Thomsen ASS, Emanuelsson F, Tendal B, Hilden J, Boutron I, Ravaud P, Brorson S. Observer bias in randomised clinical trials with binary outcomes: systematic review of trials with both blinded and non-blinded outcome assessors. BMJ. 2012;344:e1119.
Hróbjartsson A, Thomsen ASS, Emanuelsson F, Tendal B, Hilden J, Boutron I, Ravaud P, Brorson S. Observer bias in randomized clinical trials with measurement scale outcomes: a systematic review of trials with both blinded and nonblinded assessors. CMAJ. 2013;185(4):E201–11.
Lundh A, Sismondo S, Lexchin J, Busuioc OA, Bero L. Industry sponsorship and research outcome. Coch Database Syst Rev. 2012;12:MR000033.
Savovic J, Jones HE, Altman DG, Harris RJ, Juni P, Pildal J, Als-Nielsen B, Balk EM, Gluud C, Gluud LL, et al. Influence of reported study design characteristics on intervention effect estimates from randomized, controlled trials. Ann Intern Med. 2012;157(6):429–38.
Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA. 1995;273(5):408–12.
Sutton AJ, Duval SJ, Tweedie RL, Abrams KR, Jones DR. Empirical assessment of effect of publication bias on meta-analyses. BMJ. 2000;320(7249):1574–7.
Garattini S, Jakobsen JC, Wetterslev J, Bertele’ V, Banzi R, Rath A, Neugebauer EAM, Laville M, Masson Y, Hivert V et al. Evidence-based clinical practice: Overview of threats to the validity of evidence and how to minimise them. Eur J Internal Med. 2016;32:13-21.
Krogh J, Hjorthoj CR, Jakobsen JC, Lindschou J, Kessing LV, Nordentoft M, Gluud C. DEPERROR: Risks of systematic errors in drug and non-drug randomized clinical trials assessing intervention effects in patients with unipolar depression. J Affect Disord. 2015;179:121–7.
Ragland DR. Dichotomizing continuous outcome variables: dependence of the magnitude of association and statistical power on the cutpoint. Epidemiol. 1992;3(5):434–40.
Dawson NV, Weiss R: Dichotomizing continuous variables in statistical analysis. A practice to avoid. Med Decis Making 2012, 32(225).
Altman DG, Royston P. The cost of dichotomising continuous variables. BMJ. 2006;332(7549):1080.
Kirsch I, Moncrieff J. Clinical trials and the response rate illusion. Contemp Clin Trials. 2007;28(4):348–51.
Leucht S, Hierl S, Kissling W, Dold M, Davis JM. Putting the efficacy of psychiatric and general medicine medication into perspective: review of meta-analyses. Br J Psychiatry. 2012;200(2):97–106.
Brok J, Thorlund K, Wetterslev J, Gluud C. Apparently conclusive meta-analyses may be inconclusive--Trial sequential analysis adjustment of random error risk due to repetitive testing of accumulating data in apparently conclusive neonatal meta-analyses. Int J Epidemiol. 2009;38(1):287–98.
Savović J, Jones H, Altman D, Harris R, Juni P, Pildal J, Nielsen B, Balk E, Gluud C, Gluud L, et al. Influence of reported study design characteristics on intervention effect estimates from randomized controlled trials: combined analysis of meta-epidemiologic studies. Health Technol Assess. 2012;16(35):1–82.
Andrews P, Thomson JA, Amstadter A, Neale M. Primum Non Nocere: An Evolutionary Analysis of Whether Antidepressants Do More Harm than Good. Front Psychol. 2012, 3(117).
Gøtzsche PC: Deadly Medicines and Organised Crime: How Big Pharma Has Corrupted Healthcare: Radcliffe Medical Press ltd; 2013.
Cipriani A, Furukawa TA, Salanti G, Geddes JR, Higgins JP, Churchill R, Watanabe N, Nakagawa A, Omori IM, McGuire H, et al. Comparative efficacy and acceptability of 12 new-generation antidepressants: a multiple-treatments meta-analysis. Lancet. 2009;373(9665):746–58.
Acknowledgements
We thank Jørn Wetterslev and the peer reviewers for invaluable comments and suggestions.
The work was supported by The Copenhagen Trial Unit, Centre for Clinical Intervention Research, in Denmark.
Funding
The Copenhagen Trial Unit, Centre for Clinical Intervention Research, Rigshospitalet, Copenhagen, Denmark, provided economic support for JCJ, KKK, JL and CG. Otherwise, we have received no funding.
Availability of data and materials
We have included all data in the review publication and in the additional files, but if additional information is needed Janus Christian Jakobsen can always be contacted (jcj@ctu.dk).
Authors’ contributions
JCJ wrote the first draft and had full access to all the data in the review and takes responsibility for the integrity of the data and the accuracy of the data analysis. Selection of trials and data extraction: KKK, AS, SGH, SES, KLM, MI, MBB, IJP, JK, SLK, SE, AT, and JCJ. Disagreements were resolved by discussion or through arbitration by JCJ. Statistical analyses: JCJ, KKK, and JL. JCJ, JL and CG contributed to the development of the protocol and the review. All authors contributed to and have approved the manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional information
An erratum to this article is available at http://dx.doi.org/10.1186/s12888-017-1311-5.
Additional files
Additional file 1:
Search strategies. (DOC 41 kb)
Additional file 2:
List of excluded trials. (TXT 2726 kb)
Additional file 3: Table S1.
Trial characteristics. (XLS 80 kb)
Additional file 4: Figure S1.
Subgroup analysis of for profit bias. (PNG 56 kb)
Additional file 5:
Summary of findings table. (DOCX 72 kb)
Additional file 6:
Summary of the 20 Most Common Adverse Events in the Included Trials. (PDF 37 kb)
Additional file 7: Figure S3.
Meta-analysis of abnormal ejaculation. (PDF 203 kb)
Additional file 8: Figure S4.
Meta-analysis of tremor. (PDF 240 kb)
Additional file 9: Figure S5.
Meta-analysis of anorexia. (PDF 194 kb)
Additional file 10: Figure S6.
Meta-analysis of nausea. (PDF 579 kb)
Additional file 11: Figure S7.
Meta-analysis of somnolence. (PDF 459 kb)
Additional file 12: Figure S8.
Meta-analysis of sweating. (PDF 283 kb)
Additional file 13: Figure S9.
Meta-analysis of asthenia. (PDF 234 kb)
Additional file 14: Figure S10.
Meta-analysis of diarrhoea. (PDF 475 kb)
Additional file 15: Figure S11.
Meta-analysis of constipation. (PDF 369 kb)
Additional file 16: Figure S12.
Meta-analysis of insomnia. (PDF 538 kb)
Additional file 17: Figure S13.
Meta-analysis of dizziness. (PDF 406 kb)
Additional file 18 Figure S14.
Meta-analysis of dry mouth. (PDF 542 kb)
Additional file 19: Figure S15.
Meta-analysis of libido decreased. (PDF 136 kb)
Additional file 20: Figure S16.
Meta-analysis of sexual dysfunction. (PDF 124 kb)
Additional file 21: Figure S17.
Meta-analysis of appetite decreased. (PDF 123 kb)
Additional file 22: Figure S18.
Meta-analysis of fatigue. (PDF 239 kb)
Additional file 23: Figure S19.
Meta-analysis of vomiting or stomach upset. (PDF 190 kb)
Additional file 24: Figure S20.
Meta-analysis of flu syndrome. (PDF 130 kb)
Additional file 25: Figure S21.
Meta-analysis of drowsiness. (PDF 104 kb)
Additional file 26: Figure S22.
Meta-analysis of blurred or abnormal vision. (PDF 166 kb)
Additional file 27: Figure S23.
Meta-analysis of nervousness. (PDF 224 kb)
Additional file 28: Figure S24.
Meta-analysis of back pain. (PDF 153 kb)
Additional file 29: Figure S25.
Meta-analysis of headache.
Additional file 30: Figure S26
Meta-analysis of dyspepsia. (PDF 226 kb)
Additional file 31: Figure S27
Meta-analysis of weight loss. (PDF 97 kb)
Additional file 32: Figure S2
Meta-analysis of no response. (PNG 105 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Jakobsen, J.C., Katakam, K.K., Schou, A. et al. Selective serotonin reuptake inhibitors versus placebo in patients with major depressive disorder. A systematic review with meta-analysis and Trial Sequential Analysis. BMC Psychiatry 17, 58 (2017). https://doi.org/10.1186/s12888-016-1173-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12888-016-1173-2