
Abstract
Background
As a rare mitochondrial disorder, the pyruvate dehydrogenase complex (PDC) deficiency is a rare inborn disease characterized with glucose metabolism defects, which leads to neurological dysfunction, serum lactic acid buildup and a resultant trend of metabolic acidosis. Although the ketogenic diet (KD) is the first-line treatment for PDC deficiency, there is currently no widely accepted consensus on specific implementation of KD for this condition. Due to the combined effect of pre-existing hyperlactacidemia and KD-induced ketoacidosis that can further exacerbate metabolic disturbances, maintaining metabolic homeostasis should be prioritized during the implementation of KD.
Case presentation
Herein, the authors present a 6-year-old boy with lactic acidosis, ataxia, hypotonia and neuromotor development retardation. The KD was started after the patient was diagnosed with PDC deficiency based on genetic testing. The initiation with classic KD resulted in severe non-diabetic ketoacidosis with elevated anion gap, which was promptly alleviated by dextrose supplementation and dietary modification to a less-restrictive KD. Long-term supervision demonstrated the efficacy of a modified KD in improving both clinical course and metabolic acidosis of the patient.
Conclusions
This rare case adds to the limited evidence of KD application in PDC deficiency, and provides valuable insights into the importance of reasonably lowering the ketogenic ratio of KD at the start of treatment to reduce the risk of metabolic acidosis.
Similar content being viewed by others
Background
Pyruvate dehydrogenase complex (PDC) deficiency is a rare mitochondrial disorder, with genetic mutations resulting in PDC malfunction or deficiency and consequent glucose metabolism failure. The pyruvate metabolism and its conversion into acetyl-coenzyme A through the citric acid cycle might be impeded by PDC deficiency, leading to an abnormal buildup of lactate, which is alternative metabolic products of pyruvate [1]. Approximately 400 cases have been reported to date since the disease was first described in 1970 [2], but the incidence of this rare condition still remains unknown. It primarily presents with clinical manifestations of lactic acidosis, neuroanatomic lesions, ataxia, hypotonia, epilepsy and developmental delay, with many patients expiring at an early age [3].
The ketogenic diet (KD), which was initially prescribed for intractable epilepsy to reduce neuron excitability and seizure since 1921 [4, 5], was introduced as the primary therapy for PDC deficiency since 1976 [6]. With substitution of carbohydrate with fat as the main energy substrate, the KD was conducive to circumvent defects of glucose oxidation and thereby mitigate related metabolic and neurological impairments [1].
However, there is currently no consensus on the specific therapeutic approaches for implementing a KD in such patients. For patients with mitochondrial diseases who are prone to energy metabolism decompensation, ensuring stable homeostasis while starting the ketogenic diet is a crucial safety concern. Herein, we report a 6-year-old boy with PDC deficiency, presenting with hypotonia, ataxia, lactic acidosis and retardation of motor development. The initiation of classical KD was associated with clinical and biochemical amelioration, but meanwhile, yielded a metabolic acidosis which was promptly minimized by dietary adjustment with decreased ketogenic ratio. The case report adds to the limited evidence of KD application in this rare condition. What is more, to the best of our knowledge, it first indicated that initiating treatment with the classical ketogenic diet in patients with mitochondrial disorders might exacerbate metabolic acidosis. It offers valuable insights into the careful consideration needed when determining the appropriate implementation of KD management in such patients.
Case report
A 6-year-old boy was referred to Peking Union Medical College Hospital due to motor developmental delay. The boy was born preterm by caesarean section at 36 weeks of gestation, after s pregnancy complicated with maternal pregnancy-induced hypertension. The Apgar score at birth was unclear and the birth weight was 2, 200 g. The symptoms of hypotonia, poor primitive reflexes, tachypnea, lethargy and cyanosis of skin were observed at birth and the boy was kept in hospital for intensive care, further oxygen therapy, and nutritional support for 13 days. The presentation of hypotonia and lethargy persisted thereafter, with exacerbation of symptoms by even low-intensity exertion or in febrile episodes. A sustained decrease in exercise tolerance and motor skill retardation were reported. He began to lift head at 4 months, sit without support at 8 months, stand with support at 14 months, respectively. With a staggering gait, he was still in need of other one’s support to walk at the age of 4. The attack of epileptic seizure was denied, while convulsion spasm with staring spells occurred occasionally in feverish episodes. The patient was then admitted into our hospital for rapidly progressive muscular hypotonia. The parents were nonconsanguineous and no family history of neurological or metabolic disease was found.
On physical examination, the patient was somnolent and lethargic. He was 115.5 cm in height and weighed 21.2 kg. There was a generalized muscular hypotonia and the myotatic reflexes could not be elicited. An abnormal wide-based and unsteady gait was observed when he tried to walk. He had difficulty performing rapid alternating movements, finger-to-nose test and heel-to-shin test, and had a positive Romberg sign. The pathologic reflexes were not elicited.
His metabolic profile was unremarkable, except for a condition of metabolic acidosis with respiratory compensation (pH 7.416, HCO3 17.80mmol/L, base excess (BE) -5.20mmol/L, PCO2 28.3mmHg, PO2 62mmHg, Cl- 110mmol/L, Na + 148.3mmol/L), which could be exacerbated by exercise (pH 7.417, HCO3 17.30mmol/L, BE -5.50mmol/L, PCO2 27.5mmHg, PO2 66mmHg). The urinary organic acid analysis indicated slightly elevated urinary excretion of pyruvic acid (45.57mmol/mol creatinine, reference range 0 to 26 mmol/mol creatinine). The lactic acid remained within normal reference range (1.68mmol/L (0.4-2.0mmol/L)) at rest, but increased obviously to 5.58 ∼ 6.21mmol/L even with low-intensity exercise.
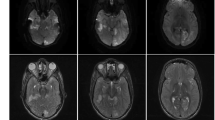
The electromyography of limbs showed decreased electrical activity in response to the muscle stimulation, suggestive of neurogenic changes. Brain magnetic resonance imaging (MRI) showed bilateral T2 hyperintensities of the globus pallidus, and lesions in periventricular tissue and left subfrontal cortex (Fig. 1). For evaluation of intelligence quotient, the patient scored 85 points by Wechsler Preschool and Primary Scale of Intelligence (Fourth Edition) (WPPSI-IV). In terms of assessment for exercise tolerance and endurance, the 6-minute walking distance (6MWD) was 216 m, compared with a reference value of 494 m for healthy boys at the same age. The genomic sequencing confirmed a hemizygote mutation: NM_000284.3 (PDHA1): c.214 C> T, p.R72C. This variant was determined to be pathogenic according to ACMG Guidelines, 1993 [PMID: 8504309] [7]. The accession number is RCV000011627.8 from the ClinVar database [8]. Combined with the results of genetic testing, a comprehensive diagnostic workup confirmed the diagnosis of PDC deficiency.

Brain magnetic resonance imaging of the patient showed bilateral T2 hyperintensities of the globus pallidus, and lesions in periventricular tissue and left subfrontal cortex
At the time of diagnosis, supplementation of the thiamine (30 mg/day) was initiated. Following 24 h of fasting (clear water allowed) period, the classic ketogenic diet (KD) was started with a ketogenic ratio of 3 ∼ 4:1. To meet the total energy expenditure, daily intake of 1400 kcal was provided via the KD, containing 85–90% fat, 5% carbohydrate and 5–10% protein (Fig. 2). Except for mild nausea without vomiting, gastrointestinal discomfort rarely occurred.
Biochemical indicators were monitored. The ketone concentration in urine dramatically increased from negative to above the detection upper limit (7.8mmol/L) after the commencement of KD and remained at this level. A metabolic acidosis aggravation (pH7.297, base excess (BE) -11.30mmol/L, anion gap (AG) 22.3, cHCO3 13.50mmol/L, PCO2 28.2mmHg, PO2 86mmHg) was reported in the fifth day post KD intervention, along with transient hypoglycemia (blood glucose 2.8mmol/L). The urine sample from that day, even when diluted fourfold by the laboratory, still indicated urine ketones ≥ 7.8 mmol/L, suggesting that the actual urine ketone concentration might be greater than 31.2 mmol/L. Given the relatively stable level of serum lactic acid (1.26 mmol/L), ketosis is the primary cause of acidosis (Table 1; Fig. 3).
Oral supplementation of 30–60 ml of dextrose 10% was provided immediately to ameliorate the ketoacidosis, followed by a modification of the classical KD to a less-restrictive variation. The patient was eventually suggested following the modified Atkins diet (with the ketogenic ratio of 0.8-1: 1), with the macronutrient composition of 60–65% fat, 10–20% carbohydrate and 20–25% protein (Fig. 2). Subsequent metabolic profile monitoring revealed alleviated metabolic acidosis (pH 7.381, BE -6.20mmol/L, AG 16.5, cHCO3 17.50mmol/L, PCO2 30.2mmHg, PO2 82mmHg, Cl- 109mmol/L, K + 4.27mmol/L, Na + 143.0mmol/L) in the sixth day post KD intervention. Follow-up assessments showed a less negative BE, decreased AG, and elevated HCO3-, indicating remaining improvement in metabolic acidosis. (Fig. 3).

The specific regimen of the ketogenic diet intervention. (A) The intervention initiation with a classic ketogenic diet (with a ketogenic ratio of 3 ∼ 4:1) providing 1400 kcal/d. (B) The subsequent change to a modified Atkins diet (with the ketogenic ratio of 0.8-1: 1) providing 1400 kcal/d

Change of the ABG following the KD implementation. A decease in pH, cHCO3, BE and an elevation in AG was associated with the classic KD initiation in 5 days; the metabolic acidosis was ameliorated promptly by small amount of glucose intake and a long-term remission of the metabolic disturbance was maintained subsequently on modified Atkins diet with or without MCT replacement
In 2 weeks after KD introduction, the boy had considerable improvement in symptoms of weakness, ataxia, lethargy and generalized hypotonia. In regard to motor improvements, he also showed enhanced exercise capacity (walking without support) and increased tolerance to physical activity, with decreased serum lactate. After discharge, the patient was maintained on the modified KD for a long term. After 6 months on KD, to improve compliance, the medium chain triglyceride (MCT) oil was substituted for 50% of conventional cooking oil, while the dietary macronutrient composition still remained unchanged. After keeping the KD for 9 months, he was presented with obvious motor development (walking, jumping and jogging independently), increased exercise endurance, and even was able to go to a primary school. The reevaluation showed acceptable ketosis, improved physical activity tolerance and diminished lactic acidosis, normal growth and development, and slightly elevated serum triglyceride and cholesterol (Table 2). The ABG was monitored till 20 months on the KD, and showed a trend of remission in metabolic acidosis (Fig. 3).
Discussion
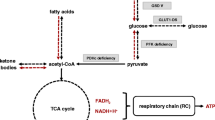
The pyruvate dehydrogenase complex (PDC) is crucial for glucose metabolism, linking glycolysis to the citric acid cycle by converting pyruvate to acetyl-coenzyme A [9]. Defects in any PDC enzymatic subunits (pyruvate dehydrogenase (E1) (including 2 α and 2 β subunits), dihydrolipoamide transacetylase (E2), dihydrolipoamide dehydrogenase (E3)) or the E3 binding protein can lead to PDC deficiency and impair mitochondrial function, glucose metabolism, and ATP production [3]. The most common cause is mutations in the X-linked PDHA1 gene, affecting the E1-α subunit, as seen in this case. It results in poor cellular energy failure, increased lactate and pyruvate levels, and progressive neurological and neuromuscular deterioration. Symptoms include hypotonia, lethargy, ataxia, tremors, seizures, developmental delay, and mental decline [3].
The ketogenic diet (KD), a high-fat, low-carbohydrate regimen, bypasses the PDC by providing fat as an alternative substrate for the citric acid cycle, thereby enhancing mitochondrial ATP production and reducing glycolysis [5]. Since the first application of KD on two boys with PDC deficiency in 1976 [6], a growing number of established evidence has emerged advocating its utility in this rare inborn metabolic disorder [10, 11]. Prior reports and case series studies has demonstrated that the KD improved life expectancy, mental development, clinical symptoms, lactate levels, and neurocognitive functions in PDC deficiency, with over 90% of patients responding positively [1, 3, 9, 12,13,14,15,16]. Based on the evidence, institutions like Johns Hopkins Hospital, the Practice Committee of the Child Neurology Society, and the International Ketogenic Diet Study Group have recommended KD as a first-line therapy for PDC deficiency, advocating for its early implementation upon diagnosis [17, 18].
However, there is no detailed consensus on how to implement the KD for PDC deficiency. Considering that PDC deficiency is a type of energy metabolism disorder, many researchers prefer to initiate KD with a lower ketogenic ratio and relatively higher carbohydrate intake, with the ratio gradually increased based on patient tolerance [12]. Meanwhile, some other studies suggest that it is possible to commence the classical KD directly with a higher ketogenic ratio of 3–4: 1 [19].
In this case, the boy with PDC deficiency underwent fasting induction followed by a classic ketogenic diet with a ketogenic ratio of 3 ∼ 4:1 and developed worsening metabolic acidosis within five days of treatment. The worsening metabolic acidosis was accompanied by a further decrease in BE, an elevated AG and a significant increase in urinary ketones, while blood lactate levels remained unchanged. Therefore, ketosis was identified as the primary cause of the exacerbation of acidosis.
Non-diabetic ketoacidosis refers to a metabolic condition where excessive production of ketone bodies leads to acidosis in the absence of diabetes mellitus. Certain inborn errors of metabolism such as ketolytic disorders are potential causes of this condition. These include monocarboxylase transporter 1 deficiency [20, 21], beta-ketothiolase deficiency [22], or Succinyl-CoA:3-ketoacid CoA transferase deficiency [23]. Non-diabetic ketoacidosis can be caused as well by the application of KD, extraordinary starvation or alcoholism, although it is rarely reported in these contexts [24,25,26]. For patients receiving KD for the treatment of PDC deficiency, this severe complication remains infrequent, with only one case briefly mentioning transient and mild ketoacidosis following KD in a 1-year-old boy with PDC deficiency [9].
The mechanism triggering KD-induced ketoacidosis has been shed light on by previous literature. When people get started on the KD, the fatty acids are mainly utilized as alternate fuel sources and undergo β-oxidation to produce acetyl CoA [27]. Under condition of low carbohydrate intake, the acetyl CoA is converted to ketone bodies, including acetoacetate, beta-hydroxybutyrate and acetone. Maintaining an adequate and sustained state of ketosis is of pivotal importance for therapeutic effects of KD [1, 12]. Secretion of insulin and glucagon in response to ketonemia can regulate formation and metabolism of ketone bodies, and prevent abnormally high levels of them in blood [28, 29]. On most occasions, the ketosis-associated acidemia was low grade, well tolerated and not clinically significant, with mild decrease in blood pH and plasma bicarbonate and increase in anion gap [27, 30, 31]. However, in rare cases, massive overproduction of ketone bodies overwhelms the acid buffer system of the body, giving rise to significant metabolic derangement. The severe KD-induced ketoacidosis has been observed in some isolated cases who maintained on KD for weight management or epilepsy control [24, 32,33,34,35]. Ullah et al. [24] summarized possible precipitating factors for this uncommon condition, including pregnancy, lactation, asthma, pneumonia, hyperthyroidism, history of bariatric surgery, and simultaneous salicylate use, all of which might make people vulnerable to ketoacidosis when under the KD treatment.
In the context of energy metabolism disorders, including PDC deficiency, the combination of pre-existing hyperlactacidemia from the primary disease and KD-induced ketosis can make individuals more susceptible to metabolic disturbances during KD treatment. Considering the risk of triggering ketoacidosis due to energy deficiency, very low carbohydrate intake, and prolonged fasting in individuals with co-existent energy metabolism decompensation, it is rational to initiate KD with a lower ketogenic ratio and higher carbohydrate content, and subsequently adjust the ketogenic ratio according to the metabolic profile.
In our case, the boy’s ketoacidosis was alleviated by the oral administration of a small amount of dextrose. Subsequently, as the classic diet (with a ketogenic ratio of 3–4:1) transitioned to a modified Atkins diet (with a ketogenic ratio of 0.8-1:1), ketoacidosis did not recur, and the metabolic profiles stabilized. This enabled the patient to adhere to KD long-term, ultimately achieving striking improvement in neuromotor development and hyperlactatemia while maintaining normal growth in height and weight. Subsequent monitoring demonstrated the effectiveness of a less-restrictive KD in both relieving the metabolic acidosis and improving clinical course of the patient with PDC deficiency.
Conclusion
As a first-line treatment for PDC deficiency, there is currently no widely accepted consensus on the specific implementation of the KD. However, considering the propensity of such diseases to be associated with energy metabolism decompensation and metabolic disturbances, initiating treatment with a modified KD with a lower ketogenic ratio might reduce the risk of non-diabetic ketoacidosis and provide better safety.
Data availability
No datasets were generated or analysed during the current study.
References
Sofou K, Dahlin M, Hallböök T, Lindefeldt M, Viggedal G, Darin N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short- and long-term outcomes. J Inherit Metab Dis. 2017;40(2):237–45.
Blass JP, Avigan J, Uhlendorf BW. A defect in pyruvate decarboxylase in a child with an intermittent movement disorder. J Clin Invest. 1970;49(3):423–32.
Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol Genet Metab. 2012;106(3):385–94.
Lee PR, Kossoff EH. Dietary treatments for epilepsy: management guidelines for the general practitioner. Epilepsy Behav. 2011;21(2):115–21.
Longo R, Peri C, Cricrì D, Coppi L, Caruso D, Mitro N et al. Ketogenic Diet: a New Light shining on Old but Gold Biochemistry. Nutrients. 2019;11(10).
Falk RE, Cederbaum SD, Blass JP, Gibson GE, Kark RA, Carrel RE. Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics. 1976;58(5):713–21.
Takakubo F, Thorburn DR, Dahl HH. A four-nucleotide insertion hotspot in the X chromosome located pyruvate dehydrogenase E1 alpha gene (PDHA1). Hum Mol Genet. 1993;2(4):473–4.
NM_000284.3(PDHA1):c.1142_1145dupATCA (p.Trp383Serfs) AND Pyruvate dehydrogenase E1-alpha deficiency. 1993 https://www.ncbi.nlm.nih.gov/clinvar/25777887/
Wijburg FA, Barth PG, Bindoff LA, Birch-Machin MA, van der Blij JF, Ruitenbeek W, et al. Leigh syndrome associated with a deficiency of the pyruvate dehydrogenase complex: results of treatment with a ketogenic diet. Neuropediatrics. 1992;23(3):147–52.
Scholl-Bürgi S, Höller A, Pichler K, Michel M, Haberlandt E, Karall D. Ketogenic diets in patients with inherited metabolic disorders. J Inherit Metab Dis. 2015;38(4):765–73.
Kraeuter AK, Guest PC, Sarnyai Z. The therapeutic potential of ketogenic Diet throughout Life: focus on metabolic, neurodevelopmental and neurodegenerative disorders. Adv Exp Med Biol. 2019;1178:77–101.
Wexler ID, Hemalatha SG, McConnell J, Buist NR, Dahl HH, Berry SA, et al. Outcome of pyruvate dehydrogenase deficiency treated with ketogenic diets. Studies in patients with identical mutations. Neurology. 1997;49(6):1655–61.
El-Gharbawy AH, Boney A, Young SP, Kishnani PS. Follow-up of a child with pyruvate dehydrogenase deficiency on a less restrictive ketogenic diet. Mol Genet Metab. 2011;102(2):214–5.
Di Pisa V, Cecconi I, Gentile V, Di Pietro E, Marchiani V, Verrotti A, et al. Case report of pyruvate dehydrogenase deficiency with unusual increase of fats during ketogenic diet treatment. J Child Neurol. 2012;27(12):1593–6.
Chida R, Shimura M, Nishimata S, Kashiwagi Y, Kawashima H. Efficacy of ketogenic diet for pyruvate dehydrogenase complex deficiency. Pediatr Int. 2018;60(11):1041–2.
DeBrosse SD, Okajima K, Zhang S, Nakouzi G, Schmotzer CL, Lusk-Kopp M, et al. Spectrum of neurological and survival outcomes in pyruvate dehydrogenase complex (PDC) deficiency: lack of correlation with genotype. Mol Genet Metab. 2012;107(3):394–402.
Kossoff EH, Zupec-Kania BA, Auvin S, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. Optimal clinical management of children receiving dietary therapies for epilepsy: updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open. 2018;3(2):175–92.
Kossoff EH, Zupec-Kania BA, Amark PE, Ballaban-Gil KR, Christina Bergqvist AG, Blackford R, et al. Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group. Epilepsia. 2009;50(2):304–17.
Zweers H, van Wegberg AMJ, Janssen MCH, Wortmann SB. Ketogenic diet for mitochondrial disease: a systematic review on efficacy and safety. Orphanet J Rare Dis. 2021;16(1):295.
van Hasselt PM, Ferdinandusse S, Monroe GR, Ruiter JP, Turkenburg M, Geerlings MJ, et al. Monocarboxylate transporter 1 deficiency and ketone utilization. N Engl J Med. 2014;371(20):1900–7.
Bozacı AE, Ünal AT. Rare cause of ketolysis: Monocarboxylate transporter 1 deficiency. Turk J Pediatr. 2022;64(4):741–6.
Grünert SC, Sass JO. 2-methylacetoacetyl-coenzyme A thiolase (beta-ketothiolase) deficiency: one disease - two pathways. Orphanet J Rare Dis. 2020;15(1):106.
Grünert SC, Foster W, Schumann A, Lund A, Pontes C, Roloff S, et al. Succinyl-CoA:3-oxoacid coenzyme a transferase (SCOT) deficiency: a rare and potentially fatal metabolic disease. Biochimie. 2021;183:55–62.
Ullah W, Hamid M, Mohammad Ammar Abdullah H, Ur Rashid M, Inayat F. Another D in MUDPILES? A review of Diet-Associated nondiabetic ketoacidosis. J Investig Med High Impact Case Rep. 2018;6:2324709618796261.
Goswami JN, Sharma S. Current perspectives on the role of the ketogenic Diet in Epilepsy Management. Neuropsychiatr Dis Treat. 2019;15:3273–85.
Ułamek-Kozioł M, Czuczwar SJ, Januszewski S, Pluta R. Ketogenic Diet and Epilepsy. Nutrients. 2019;11(10).
Gomez-Arbelaez D, Crujeiras AB, Castro AI, Goday A, Mas-Lorenzo A, Bellon A, et al. Acid-base safety during the course of a very low-calorie-ketogenic diet. Endocrine. 2017;58(1):81–90.
Alberti KG, Johnston DG, Gill A, Barnes AJ, Orskov H. Hormonal regulation of ketone-body metabolism in man. Biochem Soc Symp. 1978;43:163–82.
Manninen AH. Metabolic effects of the very-low-carbohydrate diets: misunderstood villains of human metabolism. J Int Soc Sports Nutr. 2004;1(2):7–11.
Yancy WS Jr., Olsen MK, Dudley T, Westman EC. Acid-base analysis of individuals following two weight loss diets. Eur J Clin Nutr. 2007;61(12):1416–22.
Francis BA, Fillenworth J, Gorelick P, Karanec K, Tanner A. The feasibility, Safety and Effectiveness of a ketogenic Diet for Refractory Status Epilepticus in adults in the Intensive Care Unit. Neurocrit Care. 2019;30(3):652–7.
von Geijer L, Ekelund M. Ketoacidosis associated with low-carbohydrate diet in a non-diabetic lactating woman: a case report. J Med Case Rep. 2015;9:224.
Chen TY, Smith W, Rosenstock JL, Lessnau KD. A life-threatening complication of Atkins diet. Lancet. 2006;367(9514):958.
Shah P, Isley WL. Ketoacidosis during a low-carbohydrate diet. N Engl J Med. 2006;354(1):97–8.
Basnet S, Tachamo N, Nazir S, Dhital R, Jehangir A, Donato A. Severe anion gap metabolic acidosis associated with initiation of a very low-carbohydrate diet. J Community Hosp Intern Med Perspect. 2019;9(2):165–7.
Acknowledgements
Not applicable.
Funding
This study was supported by the National High Level Hospital Clinical Research Funding (2022-PUMCH-A-146).
Ethics declarations
Ethical approval
The case study was approved by the Ethics Committee of Peking Union Medical College Hospital (Ethical Approval Number: ZS-1101) and written informed consent was obtained from the father of this 6-year-old patient in the case.
Consent for publication
The written informed consent for publication was obtained from the father of this patient in the case.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, R., Ma, M., Chen, W. et al. Classic ketogenic diet-induced ketoacidosis in the treatment of pyruvate dehydrogenase deficiency: a case report and literature review. BMC Pediatr 24, 603 (2024). https://doi.org/10.1186/s12887-024-05054-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-05054-w