
Abstract
Epidermolysis
Bullosa is a rare hereditary skin condition that causes blisters. Genes encoding structural proteins at or near the dermal-epidermal junction are mutated recessively or dominantly, and this is the primary cause of EB. Herein, two Chinese boys were diagnosed with the condition, each with a different variant in a gene that serves as a reference for EB genetic counseling. Skincare significantly impacted their prognosis and quality of life.
Case presentation
Two Chinese boys, with phenotypically normal parents, have been diagnosed with distinct blister symptoms, one with Dominant Dystrophic Epidermolysis Bullosa and the other with a severe form of Epidermolysis Bullosa Simplex. The first patient had a G-to-A variant in the COL7A1 allele, at nucleotide position 6163 which was named “G2055A”. The proband is heterozygous for Dystrophic Epidermolysis Bullosa due to a COL7A1 allele with a glycine substitution at the triple helix domain. A similar variant has been discovered in his mother, indicating its potential transmission to future generations. Another patient had severe Epidermolysis Bullosa Simplex with a rare c.377T > A variant resulting in substitution of amino acid p.Leu126Arg (NM_000526.5 (c.377T > G, p.Leu126Arg) in the Keratin 14 gene. In prior literature, Keratin 14 has been associated with an excellent prognosis. However, our patient with this infrequent variant tragically died from sepsis at 21 days old. There has been a reported occurrence of the variant only once.
Conclusion
Our study reveals that Epidermolysis Bullosa patients with COL7A1 c.6163G > A and KRT14 c.377T>A variants have different clinical presentations, with dominant forms of Dystrophic EB having milder phenotypes than recessive ones. Thus, the better prognosis in the c.6163G > A patient. Furthermore, c.377T>A patient was more prone to infection than the patient with c.6163G>A gene variant. Genetic testing is crucial for identifying the specific variant responsible and improving treatment options.
Similar content being viewed by others


Background
Epidermolysis Bullosa (EB) is a group of heritable skin disorders typified by blister development resulting from structural fragility of the skin and other epithelial tissues under mild mechanical trauma [1].
The term EB was initially described in 1886, although the first satisfactory classification method was proposed by Pearson in 1962 with global prevalence rates of 1 out of 50,000 to 500,000 live births [2].
Currently, it is divided into four primary categories based on clinical and genetic characteristics namely (a) Epidermolysis bullosa simplex (EBS) (b) Junctional epidermolysis bullosa (JEB) (c) Dystrophic epidermolysis bullosa (DEB) and (d) Kindler epidermolysis bullosa [3]. The primary distinction between the four categories is the degree of skin separation and the production of blisters that follow [4].
EB can be mild blistering to life-threatening forms with clinical signs and symptoms ranging from localized to widespread blisters and skin sores throughout the body and oral cavity caused by secondary deficiencies [5]. These may complicate chronic wounds, scars, contractures, deformities, infections, or even problems affecting several internal organs such as the esophagus, the gastrointestinal and vesico-urinary tracts, as well as the extremities. This is brought by variants in genes that affect several skin protein types, including keratins and laminin 332 type VII collagen which in turn causes the skin to become fragile. So far, these variants have been identified in 16 genes, indicating the genetic diversity of EB [4, 6]. Type VII collagen (COLVII), an essential component of the anchoring fibrils, is encoded by the COLVII-A1 (Collagen-VII-A1) gene. Variants in this gene are the primary cause of the condition: epidermal keratinocytes and papillary dermal fibroblasts in the skin produce and deposit COLVII. We hereby present two cases, one with DDEB who had blisters on bilateral lower limbs and another with EBS-gen sev who presents with severe widespread blistering and nail absence at birth. We delve into how our team managed the infants’ disease course by treating the skin symptoms efficiently. Furthermore, we explore how early diagnosis through genetic testing can aid in accuracy and better outcomes.
Case series presentation
Case 1
A 3-hour full-term male baby was referred to our hospital with bilateral asymmetrical ulcers. Despite weighing 3.1 kg and being born at full term via routine delivery without any issues, the baby presented with dermatological abnormalities that covered over 8% of his body surface and were mostly localized around both legs. The left, an irregular “S” shaped defect involves the medial aspect of the thumb, plantar region, extending up to above the knee measuring about 15*10cm (Fig. 1A). The right was a C-shaped defect around the calf area covering approximately 6 by 10 cm (Fig. 1B). Easily visualized vascular structures, a thin translucent membrane covering a bright red surface, and distinct separation from the normal skin were observed. All reflex tests presented normal except for bilateral lower limbs. The unmarried 32-year-old mother had an unremarkable medical history with no complications during the whole course of her pregnancy and denies any history of chronic or genetic disease in the family. Clinical evaluations revealed a healthy baby with no other anomalies.

(A) Skin condition of the left leg on the first day of birth. (B) Skin condition of the right leg on the first day of birth
Our hospital management involved hydration support, and the use of prophylactic antibiotics, to prevent and treat infections. Non-adhesive polysilicon wound dressing was done in intervals of 3–5 days and using sterile water, and lysosome was useful in ensuring infection was controlled. The healing process started at the peripherals (Fig. 2, and 3), and complete healing was attained in the following 45 days (Fig. 4A and B). The skin around the medial malleolus and the anterior part of the ankle joint were the final areas to recover.

(A) 16 days after birth Left leg at 5th dressing change. (B) 16 days after birth Right leg at 5th dressing change

(A) 30 days after birth Left leg at 9th dressing change. (B) 30 days after birth Right leg at 9th dressing change

(A) 42 days after birth the 11th dressing is changed on the left leg before discharge. (B) 42 days after birth the 11th dressing is changed on the right leg before discharge
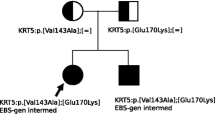
With informed parental consent for genetic testing pertinent to their child’s health status, the perilesional skin was mapped using immunofluorescence antigen during the first week of life. The results revealed a link between pathogenic variant; chr3: 48,612,789; c.6163G > A; p. Gly2055Arg in EX73/CDS73 generating an EB diagnosis and an autosomal dominant inheritance pattern whereby the child inherited this gene from the carrier mother (Fig. 6). Following a year, the baby underwent follow-up testing to look for any anomalies or developmental issues (Fig. 5A and B).
Specific blood tests, standard stool tests, and bacterial smear tests were performed. No problems or abnormalities were found during the testing process, indicating that all levels were within the anticipated normal ranges.
Additionally, (Fig. 6A, and 6B) showed first-generation sequence and genetic results of the infant with heterozygous variations in the COL7A1 gene that might connect the development of Aplasia Cutis Congenita with chromosome 3 alterations. Sanger sequencing is widely used as the first step in genetic testing because of its effectiveness in identifying gene alterations and providing accurate diagnoses.

(A) Skin condition of the left leg at the age of 1 year. (B) Skin condition of the right leg at 1 year of age

(A) Shows the patient’s Sanger sequencing technology revealing the COL7A1 gene. b illustrates the patient’s and parents’ results from genetic testing showing the COL7A1 gene. (B) A nucleotide change (c.6163G > A) in axon 73 and heterozygous variant in the COL7A1(NM_000094.3) gene was present in the affected individuals. Additionally, there is an amino acid change (p. Gly2055Arg). This genetic variation is situated at location 48,612,789 on chromosome three, in the EX73/CDS73 region. Red arrows indicated nucleotides that were mutated as seen in the Figure
Case 2
2h after birth, a spontaneously delivered male baby was brought to Jiangsu University Hospital following the discovery of a shinny red area with no skin on the hands, or feet (Fig. 7A and B) and an exfoliated epithelial-covered bolus on the buttocks with visible exudate and no purulent discharge (Fig. 8B). The child also presented with a congenital absence of fingernails and toenails. New blisters began to form from the skin defect’s margins to the medial portion. He suffered a mucosal erosion first on his mouth, which later spread to the wings of his nose (Fig. 8A). Extremities have normal muscle tone and primitive reflexes elicitation.

(A) On the first day of life, the baby’s feet feature a brilliant red, skinless region. (B) a bright red, skinless region covers the baby’s hands

(A) on day 3 illustrates a mucosal erosion that started in the mouth and spread to the nose’s wings. (B) displays a visible, exfoliated bolus covered in exudate on the buttocks upon birth
Despite being delivered at the GA of 37 + 4-week, with an Apgar score of 9/10, and having no relevant history of this disease, postnatal illness, or any other chronic illness, the patient had multiple blisters and erosions throughout the first few days of life, with a preference for places that were mechanically exposed. However, other than these abnormalities, his vital signs and general health, including weight, length, and head circumference, were normal. The 31-year-old mother denied having taken any medications or being exposed to radiation while she was pregnant. No skin anomalies or mucous membranes were visible in either parent, who appeared to be in good health. The baby is the third child of this unmarried family, the first is alive but suffers from sensory disorders and autism. However, she had a miscarriage in her second pregnancy due to unknown causes. Using serology and microbiological cultures to screen for inflammatory, autoimmune, metabolic, viral, or microbial causes of newborn blistering revealed no pathology. The findings of the liver function test, renal function test, and complete blood count were all normal. Both a cardiac echocardiogram and an abdominal ultrasound showed no abnormality. To prevent further skin damage we changed positions regularly, used non-adherent wound dressing, kept him on IV antibiotics, vitamin K to prevent bleeding, started him with nasogastric tube feeding and fluid replacement support. Gamma globulin was also administered to modulate immunity due to the elevated risk of secondary infection.
But subsequently, the baby started to deteriorate, he had hyperrespiration with oxygen saturation ranging between 90 and 94%. In addition, he developed a fever, which peaked at 37.9 to 39.6 degrees Celsius. A chest x-ray was obtained; the findings suggested pneumonia because of the small patches and bilateral lung consolidation. Based on elevated WBC and CRP levels, Table 1 validates the diagnosis of neonatal sepsis.
Consequently, we changed the baby’s medication to meropenem and kept the child on Oxygen, but the condition did not improve. Sadly, the infant died of sepsis after 21 days of hospitalization. The results of the genetic test revealed a unique variant in KRT14, known to cause EBS, c.377T > A, resulting in the amino acid substitution p.Leu126Gln on chromosome 17, consistent with the child’s symptoms. The mother of the child did not perform any genetic testing due to economic reasons, his father, however, displayed no KRT14 variant in his results (Fig. 9B). As a result, the diagnosis was established based on the clinical evaluation, the child’s, and his father’s immunofluorescence results. This patient experienced recurrent episodes of extensive bullous growth and did not considerably thrive. In the first several weeks of his life, he experienced multiple infections and poor weight gain. The most potent antibiotics were not able to improve the condition of the patient.
The father received genetic counseling, which explained the significance of these variants and recommended suitable solutions for the Child’s prognosis. (Figure 9A and B) displays the first-generation sequence and genetic testing result of the parents and patient of the child with KRT14 variant respectively.

(A) Sanger sequencing of the patient’s genome revealed the KRT14 gene. (B) The patient’s and his father’s genetic testing outcomes, displaying the KRT14 gene. NM_000526.4 is the reference sequence that illustrates the nucleotide change c.377T > A and the amino acid changes p.Leu126Gln at the gene subregion EX1/CDS1 with chromosomal location chr17:39742710; was detected in the patient but was absent in the patient’s father
Discussion
Two patients with severe forms of heterozygous hereditary EB were identified; the first patient’s clinical presentation, which comprised skin absence with bullae in both lower extremities, led to the diagnosis of both ACC and DDEB. The three most prevalent forms of DEB are “RDEB, generalized intermediate,” “DDEB, generalized,” and “RDEB, generalized severe.” “DDEB, generalized” has a favorable prognosis and exhibits decreased collagen VII expression. Blisters are localized to the site of damage and are minor. Rarely do teeth and the oral mucosa become involved [7]. The amount of disruption in Dystrophic Epidermolysis Bullosa occurs at the level of the anchoring fibrils, below the lamina densa, and causes the blisters.
The genetic test results of the our patient showed that the infant inherited the heterozygous COL7A1 c.6163G > A variant from the carrier mother, validated the diagnosis. A nucleotide substitution caused by the variant “G2055A” changed a glycine codon (GGA) into a glutamic acid codon (GAA). Although there are some existing reports of this variant from prior literature [8], new variants are still being discovered [9]. According to Diociaiuti and Randhir’s findings (Table 2), COL7A1 variants have been found to represent a dominant inheritance pattern in several cases of lower extremity aplasia cutis congenital and Bollus Dermolysis of the Newborn (BDN) [8, 10]. Our case findings, however, differ from Valeria Venti’s case, which identified a novel homozygous single-base missense variant c.6797G > T in exon 86 of the COL7A1 gene which had not been reported prior on the DEB registry [8, 11, 12]. A few variants in the COL7A1 gene are recurrent but many variants are exclusive to specific families. G2055A, found in our patient, differs from other variants, such as 5818delC, 6573 + 1G–> C, and E2857X, found in Japanese and British populations. Some studies suggest that G2043R and 425 A–> G variants are a global hotspot. Seven out of ten patients with the G2043R variant have it occurring de novo. Screening for ethnically specific variants and 425 A-G may be effective in COL7A1 variants searching [13,14,15]. Other studies indicate that in patients who are suspected of having DDEB, sequencing analysis of exons 73–75 of the COL7A1 gene can identify about 75% of the causal variants. (as well as 95% of instances if a precise DEB diagnosis is made [16]. Our patient’s better prognosis can be attributed to dominant variations of dystrophic EB, which are milder and less severe than recessive forms, according to examples previously reported in the literature. A Bruckner study found that patients with recessive dystrophic epidermolysis bullosa had the highest severity rating, with 75% classifying it as moderate to very severe (Fig. 10A and B). Further studies found frequent complications, including constipation and malnourishment having individuals with RDEB experiencing more complications than DDEB (Fig. 11) [17, 18].

A comprehending the severity of epidermolysis bullosa via patient and carers’ feedback

Comorbidities for Patients with SCC, RDEB, EB, and DEB at a 12-Month Follow-Up
The second patient’s diagnosis in our case series is EBS with c.377T > G variant, amino acid substitution p.Leu126Arg (NM_000526.5 (c.377T > G, p.Leu126Arg) on chromosome 17, which is known to cause EBS, who presented with generalized blisters and even mucosal involvement. Three main forms of EBS exist: localized (EBS-loc; OMIM number. 131,800), generalized severe (EBS-gen sev; OMIM no. 131,760), and generalized intermediate (EBS-gen intermed; OMIM no. 131,900). EBS-gen-sev is the most severe form of EBS and is caused by a variant in the keratin 5 KRT5 or KRT14. Autosomal recessive EBS is associated with both biallelic pathogenic variants in CD151 (a member of the tetraspanin superfamily), DST (Dystonin), or EXPH5 (exophilin-5) and biallelic loss-of-function variations in KRT5, KRT14, or PLEC (Plectin). Autosomal dominant EBS is associated with either a heterozygous pathogenic variant in KLHL24 or a heterozygous dominant-negative variant in KRT5, KRT14, or PLEC [19]. .
In EBS, blisters form on the basal keratinocytes, however, other research revealed that the development of epidermolysis bullosa simplex blisters was caused by the disintegration of basal and suprabasal cells. Blisters can range in severity from only affecting the hands and feet to being all over the body [20]. Skin fragility in EBS leads to non-scarring blisters and erosions brought on by slight mechanical damage. In some cases, this fragility also affects mucosal epithelia. The location of the blistering concerning the dermal-epidermal junction serves as its defining characteristic, which is distinguished from other kinds of EB or non-EB skin fragility syndromes.
The case shares similarities with one discussed by Zhang et al. (Table 2), except for congenital hyperkeratosis. Jana Kirova’s findings, discuss an EBS with muscular dystrophy (EBS-MD) along with hemorrhagic blistering, different from our patient’s [21, 22]. Autosomal dominant inheritance accounts for most cases, however, rare autosomal recessive types of EBS have been documented [23,24,25,26]. A high rate of 37% for de novo pathogenic variants in KRT14 and KRT5 has been observed in a separate study. Highly changeable CpG dinucleotides have been discovered in several codons that are more commonly impacted by these de novo variants in numerous families, however, the exact reason for the high frequency of de novo variants is yet unknown [27]. Since only one parent’s DNA was analyzed in our patient and the variant was discovered to be absent, we cannot definitively assume that this is a De novo variant. In the absence of a family history of EBS, the patient’s severe generalized EBS is explained by pathogenic variants in the KRT14 genes.
To the best of our knowledge, the KRT14 gene has been associated with multiple cases with a range of several variants in the literature, all of which have demonstrated a rather decent prognosis. On the contrary, there is just one known case of variant c.377T > A, which is the same variant our patient had, and that patient likewise had a severe form of EBS [28]. Numerous academic publications have documented sepsis-related deaths in KRT14 patients; for instance, like our patient, another EBS instance was described in which the patient also experienced septicemia [29]. However, our patient’s c.377T > A variant appears to make patients much more vulnerable to infections [30,31,32]. Sepsis, a complication associated with EB, is prevalent in various incidences, with septicemia being the most common in unspecified types of EB (Fig. 11), including the EB simplex type. In all forms of EB, oral erosions and blisters are incredibly common. Like our patient who has oral erosions and ulcers (Fig. 8a), their involvement results in neuropathic pain which makes it difficult to feed and thus, the increased likelihood of malnutrition and inability to thrive [33,34,35]. Long recognized, some kinds and subtypes of hereditary EB may be susceptible to one or more extracutaneous complications, based on case reports and small case studies seen previously. Many of them have a significant risk of morbidity, and some can even be fatal, such as in our KRT14 patients. Due to the complexity of the categorization and the large number of clinical subtypes of EB, an accurate classification of EB subtypes based only on clinical presentation is challenging. EBS-gen sev is the most severe form of EBS. It is caused by variants in the keratin 5 (KRT5) or KRT14 genes which pose a life-threatening risk to newborns. However, after infancy, particularly in late childhood and maturity, the prognosis improves. Furthermore, the prognosis for KRT14 individuals is usually bleak, some may even show significant skin abnormalities. For instance, our patient’s hands and feet were susceptible to adhesions, scarring, and contractures that might potentially hinder normal function [36,37,38].
Our approach to treating both patients involved minimizing skin fragilities by lowering the risk of secondary infections and making sure there were as few stressful situations as possible, even if conservative treatment has shown favorable results in a few cases. Prophylactic antibiotic use can lower infection risk and speed up wound healing, especially in patients with ACC and EB, as further evidenced by McCarthy and Simman et al. [39, 40]. Several other cases have shown positive outcomes [41]. By far, very few controlled trials have been conducted for the treatment of EB [42,43,44]. However, in most cases, the mainstay of treatment consists of supportive measures such as wound care, infection control, nutritional assistance, and management of any complications that may arise [45]. To facilitate this, numerous guidelines about wound care, wound cleansing, wound healing, and support for practitioners and carers have been developed to enhance the well-being of these patients [46, 47]. It is essential to managing and preventing infections, however, this may cause excruciating pain. Therefore, the psychosocial clinical practice guidelines were developed, which focused on pain management and improved the quality of life [45]. It is recommended to manage EB using the emergency care protocols [48] and a multidisciplinary approach. Thus, a group of dietitians, dermatologists, and nurses may be consulted when indicated [49, 50], since one of the primary challenges these patients face is malnutrition, as indicated in Fig. 11.
Certain complications that might develop in severe cases of EB may require surgery to manage. For instance, thoracoscopic surgery was performed on an EBS patient who experienced pneumothorax, with outstanding results [51]. However, implementing gene replacement therapy in EB patients has recently been shown to be of great benefit to improving prognosis and restoring patient’s quality of life [52,53,54]. Interestingly, some researchers have found that DDEB patients’ leg inflammation can be significantly reduced by topical tacrolimus 0.03% medication. Nonetheless, some discovered that the patient’s symptoms and quality of life improved after receiving low-dose botulinum toxin treatment [55, 56].
Clinical comparison
Due to differences in the variant sites, the two-case series of newborns with c.6163G > A variant and c.377T > A variants presented differently clinically. The COL7A1 c.6163G > A patient phenotype was less severe with the denuded skin only located on the lower limbs, while NGS analysis of the KRT14 patient revealed a c.377T > G variant having more widespread blisters that affected the hands, feet, back, buttocks, nose, and oral mucosa, which gave more room for bacterial penetration, leading to unresolved septicemia and ultimately leading to his death.
Furthermore, unlike our KRT14 patient, the COL7A1 patient had Aplasia cutis Congenita and skin absence at birth, which is a defining marker of DDEB. COL7A1 variant patients have blisters that heal with scars as seen in our patient in (Fig. 5A and B). Unlike the KRT14 variant, which are non-scarring blisters.
Blisters in COL7A1 are mostly prominent over acral sites. However, in KRT14 variants, blisters can affect the hands, feet, knees, and elbows but may additionally lead to regions of the trunk progressively becoming interspersed with hypopigmented patches.
COL7A1 induces dystrophic nails, particularly in the toenails; even though our patient’s nails were normal. KRT14 dystrophic nails can occur in the hands or feet, assuming they even have nails at all. As demonstrated by our KRT14 patient whose nails were absent.
Unlike our COL7A1 patient, our KRT14 patient had extensive palmar and plantar hyperkeratosis. While no copy number variants were found, NGS analysis of the KRT14 patient revealed a heterozygous c.377T > G variant in the KRT14 gene, which was predicted to result in the amino acid substitution p.Leu126Arg (NM_000526.5 (c.377T > G, p.Leu126Arg). Squamous Cell Carcinoma is a common complication of the COL7A1 variant, unlike the KRT14 variant where they are not commonly predisposed to this complication.
Genotype-phenotype correlation
Due to distinct genetic variants and the difference in their respective variant locations, the two patients exhibited varying phenotypic presentations and prognoses. Several academic works discuss the phenotypic severity of recessive forms of DEB in comparison to dominant ones; our patient, however, had a dominant form of DEB, which accounts for his remarkable prognosis in contrast to the patient with c.377T > G variant. One of the main causes of death for patients with KRT14 variants is sepsis; additionally, individuals who have c.377T > A variant, appear to be much more susceptible to infection than those with c.6163G > A variant.
Importance of genetic testing
EB diagnosis requires a comprehensive medical record, clinical assessment, and genetic testing due to its lack of recognition. Genetic testing aids in identifying the best treatment. To promptly counsel families on the natural history of the disease, risk of recurrence, and reproductive alternatives, experts must be knowledgeable about inheritance, age-related morbidity, and mortality linked to EB. Histopathology and molecular research are crucial for prognostication and counseling. Using genetic testing, variants in EB-related genes can be directly detected, improving clinical diagnosis to find the disease’s cause and, ultimately, a patient’s prognosis. Genetic testing is particularly advised for infertile couples or those with a history of miscarriages or stillbirths, as it can yield more thorough analysis reports and identify new variants and paternal carriers directly. In early pregnancy, genetic testing can also be performed to screen for autosomal recessive malnutrition type EB, provide scientific fertility advice, and determine the likelihood that an offspring will carry the disease.
Limitation
The main drawback of the study is that our patient’s mother, who carries the KRT14 variation, did not consent to having her DNA examined. As a result, we were unable to entirely rule out the chance that she carried the variant.
There are a few numbers of cases in our study.
Conclusion
The study reveals that dominant forms of dystrophic epidermolysis bullosa (DDEB) have milder phenotypes and are less severe than recessive forms. KRT14 variants cause sepsis, and c.377T > A variants increase infection susceptibility than c.6163G > A variant. Genetic testing is crucial for identifying the variant responsible and improving treatment options. Monitoring body temperature, WBC count, and CRP daily can aid in early diagnosis of sepsis. Our work contributes to the growing knowledge regarding COL7A1 and KRT14 variants.
Data availability
Data and materials of this case study are to be provided upon request through the corresponding author.
Abbreviations
- COL7A1:
-
Collagen Type VII Alpha 1 Chain
- KRT14:
-
Keratin 14
- KRT5:
-
Keratin5
- DDEB:
-
Dominant dystrophic Epidermolysis Bullosa
- RDEB:
-
Recessive Dystrophic Epidermolysis B
- DEB:
-
Dystrophic Epidermolysis Bullosa
- EBS:
-
Epidermolysis Bullosa Simplex
- JEB:
-
Junctional Epidermolysis Bullosa
- AD:
-
Autosomal Dominant
- AR:
-
Autosomal Recessive
- OMIM:
-
Online Mendelian Inheritance in Man (OMIM)
- CDS:
-
coding sequence
- EBS-gen-sev:
-
Epidermolysis bullosa simplex generalized severe
- EBS-loc:
-
Epidermolysis Bullosa Simplex Localized
- EBS-gen intermed:
-
Epidermolysis Bullosa Simplex generalized intermediate
- EBS-gen sev:
-
Epidermolysis Bullosa Simplex generalized severe
- PLEC:
-
Plectin
- DST:
-
Dystonin
- EXPH5:
-
Exophilin-5
- ERN-Skin:
-
European Network for Rare Skin Disorders
References
Prasad AN. Epidermolysis Bullosae. Med J Armed Forces India. 2011;67(2):165–6.
Fine JD, Bruckner-Tuderman L, Eady RAJ, Bauer EA, Bauer JW, Has C, et al. Inherited epidermolysis bullosa: updated recommendations on diagnosis and classification. J Am Acad Dermatol. 2014;70(6):1103–26.
Pfendner EG, Lucky AW. Junctional Epidermolysis Bullosa Summary Diagnosis Suggestive Findings. GeneReviews. 2018;1–30.
Has C, Bauer JW, Bodemer C, Bolling MC, Bruckner-Tuderman L, Diem A, et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183(4):614–27.
Tampoia M, Bonamonte D, Filoni A, Garofalo L, Morgese MG, Brunetti L, et al. Prevalence of specific anti-skin autoantibodies in a cohort of patients with inherited epidermolysis bullosa. Orphanet J Rare Dis. 2013;8(1):1–10.
Alharthi R, Alnahdi MA, Alharthi A, Almutairi S, Al-Khenaizan S, AlBalwi MA. Genetic Profile of Epidermolysis Bullosa cases in King Abdulaziz Medical City, Riyadh, Saudi Arabia. Front Genet. 2022;12(February):1–7.
Fozia F, Nazli R, Alrashed MM, Ghneim HK, Haq ZU, Jabeen M, et al. Detection of Novel Biallelic causative variants in COL7A1 gene by whole-exome sequencing, resulting in congenital recessive Dystrophic Epidermolysis Bullosa in three unrelated families. Diagnostics. 2022;12(7):1–12.
Yadav RS, Jayswal A, Shrestha S, Gupta SK, Paudel U. Dystrophic Epidermolysis Bullosa. J Nepal Med Association. 2018;56(213):879–82.
Liu YH, Shang X, Li ZT, Wu YM, Li Lfen, Xu XM. A novel COL7A1 gene mutation causing pretibial epidermolysis bullosa: report of a Chinese family with intra-familial phenotypical diversity. Gene. 2013;524(2):377–80.
Diociaiuti A. - Frequent Occurrence of Aplasia Cutis Congenita in Bullous Dermolysis of the Newborn [Internet]. [cited 2023 Jun 20]. Available from: https://click.endnote.com/viewer?doi=10.2340/00015555-2364&route=6
Venti V, Scalia B, Sauna A, Nasca MR, Smilari P, Praticò AD, et al. Previously unreported COL7A1 mutation in a Somali patient with Dystrophic Epidermolysis Bullosa. Mol Syndromol. 2020;10(6):332–8.
Diociaiuti A, Castiglia D, Giancristoforo S, Guerra L, Proto V, Dotta A, et al. Frequent occurrence of aplasia cutis congenita in bullous dermolysis of the newborn. Acta Derm Venereol. 2016;96(6):784–7.
Murata T, Masunaga T, Ishiko A, Shimizu H, Nishikawa T. Differences in recurrent COL7A1 mutations in dystrophic epidermolysis bullosa: ethnic-specific and worldwide recurrent mutations. Arch Dermatol Res. 2004;295(10):442–7.
Winberg J. Modulation of disease severity of dystrophic epidermolysis bullosa by a splice site mutation in combination with a missense mutation in the COL7A1 gene. Hum Mol Genet. 1997;6(7):1125–35.
Rouan F, Pulkkinen L, Uitto J, Jonkman MF, Bauer JW, Cserhalmi-Friedman PB, et al. Novel andDe Novo Glycine substitution mutations in the type VII collagen gene (COL7A1) in Dystrophic Epidermolysis Bullosa: implications for genetic counseling. J Invest Dermatology. 1998;111(6):1210–3.
Hamidi AK, Moghaddam M, Hatamnejadian N, Ebrahimi A. A novel deletion and two recurrent substitutions on type VII collagen gene in seven Iranian patients with epidermolysis bullosa. Iran J Basic Med Sci. 2016;19(8):858–62.
Bruckner AL, Losow M, Wisk J, Patel N, Reha A, Lagast H, et al. The challenges of living with and managing Epidermolysis Bullosa: insights from patients and caregivers. Orphanet J Rare Dis. 2020;15(1):1.
Feinstein JA, Bruckner AL, Chastek B, Anderson A, Roman J. Clinical characteristics, healthcare use, and annual costs among patients with dystrophic epidermolysis bullosa. Orphanet J Rare Dis. 2022;17(1).
Has C, Liu L, Bolling MC, Charlesworth AV, El Hachem M, Escámez MJ, et al. Clinical practice guidelines for laboratory diagnosis of epidermolysis bullosa. Br J Dermatol. 2020;182(3):574–92.
Kotalevskaya YY, Stepanov VA. Molecular genetic basis of epidermolysis bullosa. Vavilovskii Zhurnal Genet Selektsii. 2023;27(1):18–27.
Zhang J, Yan M, Liang J, Li M, Yao Z. A novel KRT5 mutation associated with generalized severe epidermolysis bullosa simplex in a 2-year-old Chinese boy. Exp Ther Med. 2016;12(5):2823–6.
Kyrova J, Kopeckova L, Buckova H, Mrazova L, Vesely K, Hermanova M, et al. Epidermolysis Bullosa simplex with muscular dystrophy. Review of the literature and a case report. J Dermatol Case Rep. 2016;10(3):39–48.
Khani P, Ghazi F, Zekri A, Nasri F, Behrangi E, Aghdam AM, et al. Keratins and epidermolysis bullosa simplex. Journal of Cellular Physiology. Volume 234. Wiley-Liss Inc.; 2018. pp. 289–97.
ABANMI A, JOSHI RK, ATUKORALA DN, PEDERSKN NB, KHAMIS O AL. Autosomal recessive epidermolysis bullosa simplex. A case report. Br J Dermatol. 1994;130(1):115–7.
Xu Z, Huang T, Pan M, Huang Y, Jiang Y. Case Report: recessive Dystrophic Epidermolysis Bullosa with severe esophageal stenosis: a Case Report and Literature Review. Br J Biomed Sci. 2022;79.
Sm Y, Ae U, Bch BM. Recessive Epidermolysis Bullosa simplex-A case report.
Bolling MC, Lemmink HH, Jansen GHL, Jonkman MF. Mutations in KRT5 and KRT14 cause epidermolysis bullosa simplex in 75% of the patients. Br J Dermatol. 2011;no-no.
Chong SC, Hon KL, Scaglia F, Chow CM, Fu YM, Chiu TW, et al. Severe generalized Epidermolysis Bullosa Simplex in Two Hong Kong Children due to De Novo Variants in KRT14 and KRT5. Case Rep Pediatr. 2020;2020:1–5.
Khanmohammadi S, Yousefzadeh R, Rashidan M, Hajibeglo A, Bekmaz K. Epidermolysis bullosa with clinical manifestations of sepsis and pneumonia: a case report. Int J Surg Case Rep. 2021;86.
Chern Kho Y, Rhodes LM, Robertson SJ, Su J, Varigos G, Robertson I et al. Epidemiology of Epidermolysis Bullosa in the antipodes the Australasian Epidermolysis Bullosa Registry with a focus on Herlitz Junctional Epidermolysis Bullosa. 146, Arch Dermatol. 2010.
Fine JD, Johnson LB, Weiner M, Suchindran C. Cause-specific risks of Childhood death in inherited Epidermolysis Bullosa. J Pediatr. 2008;152(2):276–e2802.
Yan Yuen W. Junctional epidermolysis bullosa. 2012.
Fine JD, Johnson LB, Weiner M, Suchindran C. Gastrointestinal complications of inherited epidermolysis bullosa: cumulative experience of the national epidermolysis bullosa registry. J Pediatr Gastroenterol Nutr. 2008;46(2):147–58.
Hubbard L, Haynes L, Sklar M, Martinez AE, Mellerio JE. The challenges of meeting nutritional requirements in children and adults with epidermolysis bullosa: Proceedings of a multidisciplinary team study day. Vol. 36, Clinical and Experimental Dermatology. 2011. p. 579–84.
Haynes L. Nutrition for children with Epidermolysis Bullosa. Dermatol Clin. 2010;28(2):289–301.
Xu Z, Huang T, Pan M, Huang Y, Jiang Y. Case Report: recessive Dystrophic Epidermolysis Bullosa with severe esophageal stenosis: a Case Report and Literature Review. Br J Biomed Sci. 2022;79(March):1–4.
Mariath LM, Santin JT, Frantz JA, Doriqui MJR, Schuler-Faccini L, Kiszewski AE. Epidermolysis Bullosa with congenital absence of skin: clinical and genetic characterization of a 23-case series. Clinical genetics. Volume 98. Denmark; 2020. pp. 99–101.
Mariath LM, Santin JT, Frantz JA, Doriqui MJR, Kiszewski AE, Schuler-Faccini L. An overview of the genetic basis of epidermolysis bullosa in Brazil: discovery of novel and recurrent disease-causing variants. Clin Genet. 2019;96(3):189–98.
Simman R. Letter to the editor: management of aplasia cutis congenital fon-scalp location (multiple letters) [1]. Br J Plast Surg. 2004;57(5):469–70.
McCarthy MA, Clarke T, Powell FC. Epidermolysis Bullosa and aplasia cutis. Int J Dermatol. 1991;30(7):481–4.
Wang Y, Song Z, Zhang L, Li N, Zhao J, Yang R, et al. Genetic analysis and prenatal diagnosis of recessive dystrophic epidermolysis bullosa caused by compound heterozygous variants of the COL7A1 gene in a Chinese family. Front Pediatr. 2022;10(November):1–7.
Uitto J, Has C, Bruckner-Tuderman L. Cell-based therapies for epidermolysis bullosa - from bench to bedside. J Dtsch Dermatol Ges. 2012;10(11):803–7.
Titeux M, Pendaries V, Hovnanian A. Gene therapy for recessive dystrophic epidermolysis bullosa. Dermatol Clin. 2010;28(2):361–6. xii.
Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363(7):629–39.
Martin K, Geuens S, Asche JK, Bodan R, Browne F, Downe A, et al. Psychosocial recommendations for the care of children and adults with epidermolysis bullosa and their family: evidence based guidelines. Orphanet Journal of Rare Diseases. Volume 14. BioMed Central Ltd.; 2019.
Korte EWH, Welponer T, Kottner J, van der Werf S, van den Akker PC, Horváth B, et al. Heterogeneity of reported outcomes in epidermolysis bullosa clinical research: a scoping review as a first step towards outcome harmonization. Br J Dermatol. 2023;189(1):80–90.
Pope E, Lara-Corrales I, Mellerio J, Martinez A, Schultz G, Burrell R, et al. A consensus approach to wound care in Epidermolysis Bullosa. J Am Acad Dermatol. 2012;67(5):904–17.
Mellerio JE, El Hachem M, Bellon N, Zambruno G, Buckova H, Autrata R, et al. Emergency management in Epidermolysis Bullosa: Consensus clinical recommendations from the European reference network for rare skin diseases. Orphanet Journal of Rare Diseases. Volume 15. BioMed Central Ltd.; 2020.
Has C, El Hachem M, Bučková H, Fischer P, Friedová M, Greco C, et al. Practical management of epidermolysis bullosa: consensus clinical position statement from the European Reference Network for Rare skin diseases. J Eur Acad Dermatol Venereol. 2021;35(12):2349–60.
El Hachem M, Zambruno G, Bourdon-Lanoy E, Ciasulli A, Buisson C, Hadj-Rabia S, et al. Multicentre consensus recommendations for skin care in inherited epidermolysis bullosa. Orphanet J Rare Dis. 2014;9:76.
Yukawa H, Makino T, Hayashi K, Date H, Honda N, Anami Y. Perioperative Management of Congenital Epidermolysis Bullosa. Ann Thorac Surg. 2023;116(1):e1–4.
Bischof J, Hierl M, Koller U. Emerging gene therapeutics for Epidermolysis Bullosa under Development. Int J Mol Sci. 2024;25(4):2243.
Niti A, Koliakos G, Michopoulou A. Stem cell therapies for Epidermolysis Bullosa Treatment. Volume 10. Bioengineering. MDPI; 2023.
Simman R. Letter to the editor: management of aplasia cutis congenital fon-scalp location (multiple letters) [1]. British Journal of Plastic Surgery. Volume 57. Churchill Livingstone; 2004. pp. 469–70.
Almaani N, Liu L, Perez A, Robson A, Mellerio JE, McGrath JA. Epidermolysis Bullosa pruriginosa in association with lichen planopilaris. Clin Exp Dermatol. 2009;34(8):e825–8.
Melendez M, Osborn L, Bowers E. Low-dose botulinum toxin improves disease activity and quality of life in epidermolysis bullosa simplex. Int J Dermatol. 2023;62(8).
Acknowledgements
Our deepest gratitude should be extended to the patients first, and to all staff of the Neonatal Department in Affiliated Hospital of Jiangsu University for their support.
Funding
No funding was used to prepare this case series and literature review.
Author information
Authors and Affiliations
Contributions
Fatma Mabrouk Ali wrote the main manuscript. Jieyu Zhou contributed to finding the case and collecting all the information about the patients. Mingyan Wang performed visual data description. Qiuxia Wang prepared the figures. Lulu Sun edited the manuscript. Mansour Maulid Mshenga counterproofs the manuscript. Hongyan Lu ensured the manuscript was accurately written and journal requirements were met.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not Applicable.
Consent for publication
Informed consent for this case series has been obtained to publish the information, images, and chart review from the parents of both patients.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ali, F.M., Zhou, J., Wang, M. et al. Epidermolysis Bullosa: Two rare case reports of COL7A1 and EBS-GEN SEV KRT14 variants with review of literature. BMC Pediatr 24, 242 (2024). https://doi.org/10.1186/s12887-024-04715-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-04715-0