
Abstract
Background
NARS2 as a member of aminoacyl-tRNA synthetases was necessary to covalently join a specific tRNA to its cognate amino acid. Biallelic variants in NARS2 were reported with disorders such as Leigh syndrome, deafness, epilepsy, and severe myopathy.
Case presentation
Detailed clinical phenotypes were collected and the NARS2 variants were discovered by whole exome sequencing and verified by Sanger sequencing. Additionally, 3D protein structure visualization was performed by UCSF Chimera. The proband in our study had early-onset status epilepticus with abnormal EEG and MRI results. She also performed global developmental delay (GDD) and myocardial dysfunction. Next-generation sequencing (NGS) and Sanger sequencing revealed compound heterozygous missense variants [NM_024678.6:exon14: c.1352G > A(p.Arg451His); c.707T > C(p.Phe236Ser)] of the NARS2 gene. The proband develops refractory epilepsy with GDD and hyperlactatemia. Unfortunately, she finally died for status seizures two months later.
Conclusion
We discovered two novel missense variants of NARS2 in a patient with early-onset status epilepticus and myocardial dysfunction. The NGS enables the patient to be clearly diagnosed as combined oxidative phosphorylation deficiency 24 (COXPD24, OMIM:616,239), and our findings expands the spectrum of gene variants in COXPD24.
Similar content being viewed by others

Introduction
Asparaginyl-tRNA synthetase 2 (Asn-RS) encoded by NARS2 is a member of the class II family of aminoacyl-tRNA synthetases (aaRSs) which play a crucial role in biosynthesis by catalyzing the ligation of asparagine to tRNA molecules [1]. This protein was first identified in 2005 [2] and contains 477 amino acids. It is expressed ubiquitously throughout the body both in humans and mice [1]. Moreover, it is expected to function as a dimer [2].
Pathogenic variants in NARS2 have been subsequently identified [3, 4] and were correlated to the combined oxidative phosphorylation deficiency 24 (COXPD24) (OMIM: 616,239) which is an autosomal recessive mitochondrial disorder that exhibits pleiotropic phenotypes. It is associated with visual and hearing abnormalities, myopathy, neurodevelopmental disorder, and mitochondrial dysfunction [5,6,7,8,9].
Here we present a further patient from a non-consanguineous family with an infantile-onset neurodegenerative disorder characterized by status epilepticus, increased serum lactic acid, and abnormal brain structure. The novel compound heterozygous variants in NARS2 [NM_024678.6: c.1352G > A(p.Arg451His); c.707T > C(p.Phe236Ser)] were identified by whole exome sequencing (WES). And our findings expand the genotype spectrum of COXPD24.
Methods
Patient
Patient with early-onset status epilepticus have been confirmed at the First hospital of Jilin University. Informed consent was provided from the families contained according to institutional guidelines. Ethics approval has been obtained by the human ethics committees of Bethune First Hospital of Jilin University. The clinical, laboratory examinations, and genetic tests were obtained for the patients.
WES
The genomic DNA isolated from the peripheral blood of our patient, her parents and brother. Exome captures were performed using the IDT xGen Exome Research Panel with paired-end read sequences generated on NovaSeq 6000 sequencing. Sequences were aligned to Human reference genome GRCh38/hg38 using the Burrows-Wheeler Aligner (BWA) [10]. The variants were then annotated through AnnoVar [11] and evaluated according to allele frequencies, pathogenicity prediction, and protein function. The pathogenicity of variants were predicted in silico for missense variants (SIFT, PolyPhen2, LRT, MutationTaster, FATHMM, CADD, REVEL) and for splice site variants (MaxEntScan, NNSplice, dbscSNV) [12]. Variants with minor allele frequency < 0.005 were selected, and were classified according to inheritance pattern. Candidate variants were finally screened according to the American College of Medical Genetics and Genomics (ACMG) [13] classification guidelines and clinical phenotypes.
The criteria for variant filtering were as follows:
-
1.
Variants located in exon and splicing (± 20 bp) region and minor allele frequency < 0.005 for genome aggregation database (gnomAD) exome_popmax, gnomAD_genome_popmax, gnomAD3_genome_AF_ Popmax, and etc. were selected.
-
2.
Missense variations predicted harmful by most commonly used software will be adopted.
-
3.
Then variants were classified according to inheritance pattern: de novo variants, autosomal recessive (AR) inheritance of homozygous variants, AR inheritance of compound heterozygous variants, X-linked inheritance (Supplementary Table 1).
Pathogenic variants related to clinical phenotypes will further be verified by Sanger sequencing. Primers were designed with Primer3 software [14]. Polymerase chain reaction (PCR) amplified products were purified and then sequenced with BigDye v3.1 (Applied Biosystems).
3D protein structure modeling
Molecular modeling analysis was performed to show the variations in protein structure. The homology models in the NARS2 protein based on the crystal structure of the Elizabethkingia Asparagine-tRNA ligase were predicted by the Swiss-Model program [15]. The human NARS2 model was downloaded in the AlphaFold dataset [16]. UCSF Chimera software was used to visualize the structures in dimmers and monomers [17].
Results
Case presentation
The patient was the second child of healthy non-consanguineous parents. This patient was born on an uneventful full-term cesarean delivery with a birth weight of 3.55 kg. However, the global developmental delay (GDD) was found in our patient with difficulty to head control, roll over, eyes following objects, and feeding in her three-month-old. Furthermore, her weight gains slowly after birth, weighing only 5.5 kg at 3 months old.
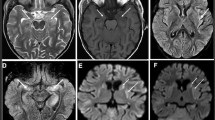
She was admitted to our hospital due to intermittent fever, seizures, eyes upward rolling and salivation when she was 3 months old. She was initially diagnosed with epilepsy and developmental delay for the abnormal electroencephalograph (EEG) and Magnetic Resonance Imaging (MRI) results. EEG showed the background rhythm slowed down and mixture multiple foci-spikes, spike waves, and sharp waves (Fig. 1A). There were frequent focal subclinical or clinical seizures arising from the left frontotemporal (Fig. 1B). An abnormal signal in the splenium of the corpus callosum was shown in MRI (Fig. 1D). Her intermittent fever was gradually controlled through anti-infection treatment. However, the status seizures failed to remission for continuous adjustment the types and dosages of antiepileptic drug.

EEG and MRI results in patient with NARS2 heterozygous variant. A EEG results showed multiple foci-spikes, spike waves, and sharp waves when patient was 3 months and 20 days old. B The patient exhibited upward rolling of the eyes and salivation, while the EEG shows synchronization with low to medium amplitude fast wave rhythm in the left frontotemporal. C The EEG presented highly irregular burst suppression patterns when patient was 4 months and 16 days old. D At the age of 3 months and 12 days, the diffusion-weighted image (DWI) revealed a small and slightly high signal shadow in the corpus callosum. while an apparent diffusion coefficient map (ADC) showed a slightly low signal intensity. E At the age of 4 months and 15 days, the T1-weighted image showed abnormalities in bilateral symmetry signal, and decreased white matter in bilateral cerebral
She also had a myocardial dysfunction with elevated myocardial creatine kinase (CK-MB 46.0U/L, ref: 0-25U/L) and B-type natriuretic peptide precursor (PRO-LPBN 209.0pg/ml, ref: 0-125pg/ml). Sodium creatine phosphate was given as nutritional myocardial therapy. Furthermore, increased serum lactic acid (5.8mmol/L, ref: 0.5-2.2mmol/L) was suspected for mitochondrial genetic disease. The brainstem auditory evoked potentials (BAEP) showed bilateral suspicious peripheral damage, combined with central damage suggesting hearing impairment. EEG and MRI were rechecked after 1 week in hospital. EEG present highly irregular with burst suppression patterns (Fig. 1C). White matter volume was reduced and bilateral symmetry signal abnormalities were shown in repeat MRI (Fig. 1E). She was admitted to the intensive care unit two times for status seizures. Twenty-six days after admission, her seizures were still frequent. Her breathing and swallowing decreased, her heart rate, blood oxygen, and various vital signs were not stable. She was discharged due to her parents’ strong request. She was still suffered from feeding difficulties and with breathing difficulty. Unfortunately, she passed away (8-month-old) at the local hospital with multiple organ failure and malnutrition 3 months later.
Identification of NARS2 variations by WES
Trio WES was subsequently performed to further investigate the etiology for the patient from a non-close relative’s family. Variants were filtered by the minor allele frequency (MAF), related phenotype and predicted damage. We listed variants as candidate pathogenic genes (Supplementary Table 1), some of which were excluded because they only explained part of the patient’s phenotype, or the inheritance pattern did not match. Two novel compound heterozygotes in NARS2 [NM_024678.6: c.1352G > A (p.Arg451His); c.707T > C (p.Phe236Ser)] were identified. These two variants have not been included in public data such as the gnom AD (https://gnomad.broadinstitute.org/gene/ENSG00000134440?dataset=gnomad_r2_1) [18, 19], and variant c.1352G > A (p.Arg451His) has a very low MAF with 0.00003184 in gnom AD_genome_ALL (Table 1). Additionally, 107 single nucleotide variations in NARS2 were recorded so far in ClinVar https://www.ncbi.nlm.nih.gov/clinvar/?term=NARS2%5Bgene%5D, and neither of the two variants in our patient were included. And both variants in our patient were predicted being damage by several prediction software (Table 1). It seems that variants in NARS2 are rare both in the disease group (Clinvar) or general population (gnom AD), and the pathogenicity needs to be further investigated.
The heterozygous variants in the patient were confirmed by Sanger sequencing and they were inherited from her parents (Fig. 2A-B). The variant c.707T > C was inherited from her father, and another variant c.1352G > A was inherited from her mother (Fig. 2B). All these two variants changed the amino acids were conserved in multiple species (Fig. 2C), may indicating their important functions. Studies of patients with NARS2 variations showed variable clinical phenotypes (Table 2). They may also be associated with additional complications as various degrees of intellectual disability, visual, hearing impairment, and developmental delay. NARS2 gene variations were identified in patients with autosomal recessive deafness and COXPD24, and most of them were missense. A schematic diagram of NARS2 variations was shown in Fig. 2D.

The variant information of NARS2. A The pedigree of this family. The proband affected with status seizures is indicated by black filled symbols and arrows. The parents who carried variants are displayed by symbols with black dots in the center. B Sanger sequencing of this family showed compound heterozygous variants c.70T > c, C.1352G > A (red box) in the proband were inherited from her parents respectively. C Variants of NARS2 in our patient located highly conserved areas based on the comparison performed among multiple species. D Domain structure and modeling of the known NARS2 variations in previous studies. The NARS2 protein contains an OB-fold nucleic acid binding domain (green) and an aminoacyl-tRNA synthetase domain (red). Three conservative motifs were shown in the structure (yellow). Variants in our study were highlighted in red font. All the compound heterozygous variations were linked by a dashed gray line
Protein modeling
To understand the molecular structures of the NARS2, comparative modeling was performed using the Swiss-Model. Due to the human Asn-RS crystallographic structure has not yet been clarified, the homology model based on Elizabethkingia sp. was used (QMEAND is Co Global 0.74) to predict and exhibited the structures of Asn-RS (Fig. 3A). All the two variants in this case were located in the aminoacyl-tRNA synthetase domain (http://pfam.xfam.org/family/PF00152) which play a crucial role in catalyzes the attachment of an amino acid to its cognate transfer RNA molecule. The variants in the dimer model were highlighted with yellow (p.Phe236Ser) and green (p.Arg451His) spheres. The changes of residues were visualization through UCSF Chimera and the stability of protein structure was predicted by mutations cut off scanning matrix (mCSM) (https://biosig.lab.uq.edu.au/mcsm/) and DUET (https://biosig.lab.uq.edu.au/duet/) (Fig. 3B, C). All the scores of mCSM method (http://biosig.unimelb.edu.au/mcsm/) and DUET server (http://biosig.unimelb.edu.au/duet/) that showed destabilizing for the residues’ changes. At the same time, the variants were highlighted in the monomer model with yellow and green as ball and stick (Fig. 3D). The ATP binding motif (motif 3) was displayed with blue spheres and the variant p.Arg451His was included indicating that the missense variant may affect the synthetase function of Asn-RS.

Protein modeling of NARS2. A The homodimer of Asn-RS in Elizabethkingia was modelled by Swiss-Model. Arg451 and Phe236 were presented by green and yellow spheres respectively. B, C The mutated residues were shown in the enlarged photos, and the predicted stability impact through mCSM and DUET was shown. D The variations in monomer predicted by AlphaFold were presented by yellow and green ball-stick. Motif 3 which was crucial in ATP binding was highlighted by blue spheres. The H bonds for Phe236 are shown in a partially enlarged view (purple lines) that are linked to Ile178 and Val176
Discussion
The aaRSs are a group of enzymes that facilitate the ligation of 20 amino acids to their molecular cognate tRNA [29]. Variations in aaRSs were reported leading to central nervous system (CNS) pathologies with epileptic encephalopathy, developmental delay, and intellectual disability [30]. NARS2 is a member of the class II family of aaRSs to catalyze the ligation of asparagine to tRNA molecules in the mitochondrion. The variant of NARS2 was first reported in two siblings with myopathy and combined complex I and IV deficiency in skeletal muscle [4]. NARS2 deficiency may cause a decrease in oxygen consumption rates and electron transport chain activities in patient fibroblasts [1]. The specific cardiac dysfunction and neonatal diabetes phenotypes are supplied in NARS2 variant individuals. On the whole, they mainly present status seizures, visual hearing disorder, and severe myopathy that was identified as the pathogenic gene of COXPD24 (OMIM:616,239).
A comprehensive review of NARS2 mutations was performed. Up to now, only 28 variants in NARS2 gene have been reported, and the exact genotype-phenotype correlation is not clear. The number of reported cases related to NARS2 deficiency has been gradually increasing [3, 5,6,7, 9, 26,27,28]. Recently, more individuals of NARS2 variants have been reported [9, 22,23,24,25]. Data from this study was compared with 28 variants in NARS2 gene published studies. Their diagnosis, phenotype, variant type, zygote type, survival outcome, and clinical finding are summarized in Table 2. Domain structure and modeling of the known NARS2 variations in previous studies in Fig. 2D.
Epileptogenesis is commonly associated with neurodegeneration and bioenergetic defects and mitochondrial dysfunction decline of energy by dysfunction of the electron transport chain leading to apoptotic neuronal death [31]. As previous studies, neurodevelopmental disorders were the main features of NARS2 deficiency. Most of the patients with NARS2 variants had focal, generalized, or myoclonic seizures and mitochondrial abnormalities such as combined complexes decreased and structurally abnormal [3]. In this study, a female infant with intermittent fever, status seizures, and GDD was described. GDD presented as difficulty in head control and roll over at her four-month-old. Status frequent focal subclinical or clinical seizures in the left frontotemporal were observed by long-term EEG monitor. Moreover, the brain structure abnormal was also detected in our patient with abnormal single and bilateral white matter atrophy in MRI. These clinical features were commonly in diseases with aaRSs gene mutations, including leukoencephalopathy with thalamus and brainstem involvement and high lactate (LTBL) cases with NARS2 variations, leukoencephalopathy with brainstem and spinal cord involvement, lactate elevation (LBSL) with Aspartyl-tRNA Synthetase 2 (DARS2) variations, and Alanyl-tRNA Synthetase 2 (AARS2)-related leukoencephalopathy [29]. It seems that there may be a shared mechanism of mitochondrial dysfunction in these disorders.
Severe myopathy was another characteristic clinical feature for cases with NARS2 variant. It is well known that mitochondrial dysfunction will affect tissues request high-energy such as brain, muscle, and heart. Patients with NARS2 deficiency usually develop muscle weakness of limbs and face muscles. Myocardial dysfunction in this case was represented with CK-MB and PRO-LPBN evaluated. Heart phenotype in patients with NARS2 deficiency was rare with mitral valve prolapse [9] and cardiac dysfunction [22], while myocardial dysfunction has been reported in other aaRSs, including AARS2 [32] and Lysyl-tRNA synthetase (KARS) [7, 33]. The reported patient with cardiac dysfunction has same phenotype with our patient and persistent elevation of serum hepatic and myocardial enzymes, but further investigation is necessary to verify whether NARS2 variants lead to cardiomyopathy.
Individuals with the same variant could exhibit different phenotypes in identical [4] or unrelated [7] families. While some clinical features with vision impairment were specifically present in some cases but not found in our patient. This may be explained by tissue specificity that consistent with other mitochondrial diseases [34]. The broad phenotypic variability of NARS2 related disease present from an infantile lethal phenotype to mild non-progressive disease. Therefore, there may be no strong association between the genetic variants and disease severity [7].
All variants observed in NARS2 were located in functional domains of NARS2 (Fig. 1D). Most of them were missense and may lead to protein dysfunction by changing the stability or interactions with other biological molecules [35]. The compound heterozygous variations of our patient in NARS2 [c.1352G > A (p.Arg451His); c.707T > C (p.Phe236Ser)] are located in the aminoacyl-tRNA synthetase domain. This domain contains three conservative motifs which are also found in other classII aaRSs. Among them, motif 3 contains strictly conserved arginine (Arg) residue that plays a crucial role in adenosine triphosphate (ATP) binding function [36]. Based on protein modeling analyses, the variant c.1352G > A; p.Arg451His (Fig. 3) changes Arg to His that is from a conserved non-aromatic to an aromatic, differently shaped, and this changing conserved Arg in motif 3 may affect ATP binding and the NARS2 function. Furthermore, another pathogenic mutation for the change of the Arg residue (from Arg to Cys) was also shown in another patient with Leigh syndrome [20]. Another variant in our patient in the 236th amino acid changes one amino acid to another that is more polar, smaller, and more flexible. It was found to have intermolecular hydrogen bonds with the 176th and 178th residues that were contained in conserved motif 1 (Fig. 3D). The crucial role of motif 1 [37] in dimerization may be affected by Phe236Ser. Meanwhile, another changed residue in 236th (from Phe to Cys) was found in patients with the infantile-onset neurodegenerative disorder [5] which explains the pathogenicity of this variation. The two NARS2 variants in our patient were predicted by mCSM and DUET software to have a stability change in the structure of the protein (Fig. 3). Unfortunately, our study was lacking in the validation of in vivo or vitro experiments. Given the patient’s ultimate demise, we will address this shortcoming in our future research.
In conclusion, we identified the novel compound heterozygous variants in an infantile-onset patient with status epilepticus and neurodegenerative disorder with final diagnosis as mitochondrial encephalomyopathy. Our study expands the genotype spectrum of COXPD24 and highlights the critical role of NARS2 in epilepsy and neurodevelopment.
Availability of data and materials
The DNA sequence data and Genetic variation data were used in our study. The data that support the findings of this study are available from the corresponding author upon reasonable request. Supplementary data to this article can be found online at https://www.ncbi.nlm.nih.gov/clinvar/variation/2663835/?oq=SCV004171024&m=NM_024678.6(NARS2):c.707T%3EC%20(p.Phe236Ser), https://www.ncbi.nlm.nih.gov/clinvar/variation/2663834/?oq=SCV004171023&m=NM_024678.6(NARS2):c.1352G%3EA%20(p.Arg451His).
Abbreviations
- GDD:
-
The global developmental delay
- NGS:
-
Next-generation sequencing
- COXPD24:
-
combined oxidative phosphorylation deficiency 24
- Asn-RS:
-
Asparaginyl-Trna synthetase
- aaRSs:
-
Aminoacyl-tRNA synthetases
- WES:
-
Whole exome sequencing
- BWA:
-
Burrows-Wheeler Aligner
- ACMG:
-
American College of Medical Genetics and Genomics
- gnomAD:
-
Genome aggregation database
- AR:
-
Autosomal recessive
- PCR:
-
Polymerase chain reaction
- EEG:
-
Electroencephalograph
- MRI:
-
Magnetic Resonance Imaging
- CK-MB:
-
Myocardial creatine kinase
- PRO-LPBN:
-
B-type natriuretic peptide precursor
- BAEP:
-
Brainstem auditory evoked potentials
- MAF:
-
Minor allele frequency
- mCSM:
-
Mutations cut off scanning matrix
- CNS:
-
Central nervous system
- LTBL:
-
Leukoencephalopathy with thalamus and brainstem involvement and high lactate
- LBSL:
-
Leukoencephalopathy with brainstem and spinal cord involvement lactate elevation
- DARS2:
-
Aspartyl-tRNA Synthetase 2
- AARS2:
-
Alanyl-tRNA Synthetase 2
- KARS:
-
Lysyl-tRNA synthetase
- Arg:
-
Arginine
- ATP:
-
Adenosine triphosphate
References
Simon M, Richard EM, Wang X, Shahzad M, Huang VH, Qaiser TA, Potluri P, Mahl SE, Davila A, Nazli S, et al. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 2015;11(3): e1005097.
Bonnefond L, Fender A, Rudinger-Thirion J, Giegé R, Florentz C, Sissler M. Toward the full set of human mitochondrial aminoacyl-tRNA synthetases: characterization of AspRS and TyrRS. Biochemistry. 2005;44(12):4805–16.
Sofou K, Kollberg G, Holmström M, Dávila M, Darin N, Gustafsson CM, Holme E, Oldfors A, Tulinius M, Asin-Cayuela J. Whole exome sequencing reveals mutations in NARS2 and PARS2, encoding the mitochondrial asparaginyl-tRNA synthetase and prolyl-tRNA synthetase, in patients with Alpers syndrome. Mol Genet Genomic Med. 2015;3(1):59–68.
Vanlander AV, Menten B, Smet J, De Meirleir L, Sante T, De Paepe B, Seneca S, Pearce SF, Powell CA, Vergult S, et al. Two siblings with homozygous pathogenic splice-site variant in mitochondrial asparaginyl-tRNA synthetase (NARS2). Hum Mutat. 2015;36(2):222–31.
Mizuguchi T, Nakashima M, Kato M, Yamada K, Okanishi T, Ekhilevitch N, Mandel H, Eran A, Toyono M, Sawaishi Y, et al. PARS2 and NARS2 mutations in infantile-onset neurodegenerative disorder. J Hum Genet. 2017;62(5):525–9.
Seaver LH, DeRoos S, Andersen NJ, Betz B, Prokop J, Lannen N, Jordan R, Rajasekaran S. Lethal NARS2-related disorder associated with rapidly progressive intractable epilepsy and global brain atrophy. Pediatr Neurol. 2018;89:26–30.
Sofou K, Kollberg G, Hedberg-Oldfors C, Oldfors A. The phenotypic variability and natural history of NARS2 associated disease. Eur J Paediatr Neurol. 2021;31:31–7.
Štěrbová K, Vlčková M, Hansíková H, Sebroňová V, Sedláčková L, Pavlíček P, Laššuthová P. Novel variants in the NARS2 gene as a cause of infantile-onset severe epilepsy leading to fatal refractory status epilepticus: case study and literature review. Neurogenetics. 2021;22(4):359–64.
Vafaee-Shahi M, Farhadi M, Razmara E, Morovvati S, Ghasemi S, Abedini SS, Bagher Z, Alizadeh R, Falah M. Novel phenotype and genotype spectrum of NARS2 and literature review of previous mutations. Ir J Med Sci. 2021;191(4):1877–90.
Abuín JM, Pichel JC, Pena TF, Amigo J. BigBWA: approaching the burrows-wheeler aligner to big data technologies. Bioinformatics. 2015;31(24):4003–5.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–24.
Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86.
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–w303.
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–9.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–43.
Collins RL, Brand H, Karczewski KJ, Zhao X, Alföldi J, Francioli LC, Khera AV, Lowther C, Gauthier LD, Wang H, et al. A structural variation reference for medical and population genetics. Nature. 2020;581(7809):444–51.
Lee JS, Yoo T, Lee M, Lee Y, Jeon E, Kim SY, Lim BC, Kim KJ, Choi M, Chae JH. Genetic heterogeneity in Leigh syndrome: highlighting treatable and novel genetic causes. Clin Genet. 2020;97(4):586–94.
Palombo F, Graziano C, Al Wardy N, Nouri N, Marconi C, Magini P, et al. Autozygosity-driven genetic diagnosis in consanguineous families from Italy and the Greater Middle East. Hum Genet. 2020;139(11):1429–41.
Zhang Y, Zhao X, Xu Y, Chen L, Li N, Yao R, Wang X, Wang J, Yu T. Study of novel NARS2 variants in patient of combined oxidative phosphorylation deficiency 24. Transl Pediatr. 2022;11(4):448–57.
Yagasaki H, Sano F, Narusawa H, Watanabe D, Kaga Y, Kobayashi K, Asano Y, Nagata M, Yonei A, Inukai T. Compound heterozygous variants of the NARS2 gene in siblings with developmental delay, epilepsy, and neonatal diabetes syndrome. Am J Med Genet A. 2022;188(8):2466–71.
Yang Z, Cao J, Song Y, Li S, Jiao Z, Ren S, Gao X, Zhang S, Liu J, Chen Y. Whole-exome sequencing identified novel variants in three Chinese Leigh syndrome pedigrees. Am J Med Genet A. 2022;188(4):1214–25.
Tanaka R, Takeguchi R, Kuroda M, Suzuki N, Makita Y, Yanagi K, Kaname T, Takahashi S. Novel NARS2 variant causing leigh syndrome with normal lactate levels. Hum Genome Var. 2022;9(1):12.
Al-Sharif F, Alsadeq H, Rozan A, Halabi MB, Badwilan H, Mohammed AA, Rahman M, Balgith T. Bilateral nonsyndromic sensorineural hearing loss caused by a NARS2 mutation. Cureus. 2022;14(11):e31467.
Cokyaman T, Cetin H, Dogan D, Silan F. A new entity in the NARS2 variant: the first reported case of type 1 diabetes mellitus associated with the phenotype. J Trop Pediatr. 2022;69(1):fmac108.
Hu W, Fang H, Peng Y, Li L, Guo D, Tang J, Yi J, Liu Q, Qin W, Wu L, et al. Clinical and genetic analyses of premature mitochondrial encephalopathy with epilepsia partialis continua caused by novel biallelic NARS2 mutations. Front Neurosci. 2022;16: 1076183.
Fine AS, Nemeth CL, Kaufman ML, Fatemi A. Mitochondrial aminoacyl-tRNA synthetase disorders: an emerging group of developmental disorders of myelination. J Neurodev Disord. 2019;11(1):29.
Diodato D, Ghezzi D, Tiranti V. The mitochondrial aminoacyl tRNA synthetases: genes and syndromes. Int J Cell Biol. 2014;2014:787956.
Singh S, Singh TG, Rehni AK, Sharma V, Singh M, Kaur R. Reviving mitochondrial bioenergetics: a relevant approach in epilepsy. Mitochondrion. 2021;58:213–26.
Götz A, Tyynismaa H, Euro L, Ellonen P, Hyötyläinen T, Ojala T, Hämäläinen RH, Tommiska J, Raivio T, Oresic M, et al. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88(5):635–42.
Ardissone A, Tonduti D, Legati A, Lamantea E, Barone R, Dorboz I, Boespflug-Tanguy O, Nebbia G, Maggioni M, Garavaglia B, et al. KARS-related diseases: progressive leukoencephalopathy with brainstem and spinal cord calcifications as new phenotype and a review of literature. Orphanet J Rare Dis. 2018;13(1):45.
Seneca S, Goemans N, Van Coster R, Givron P, Reybrouck T, Sciot R, Meulemans A, Smet J, Van Hove JL. A mitochondrial tRNA aspartate mutation causing isolated mitochondrial myopathy. Am J Med Genet A. 2005;137(2):170–5.
Chen Y, Lu H, Zhang N, Zhu Z, Wang S, Li M. PremPS: Predicting the impact of missense mutations on protein stability. PLoS Comput Biol. 2020;16(12): e1008543.
Perona JJ, Hadd A. Structural diversity and protein engineering of the aminoacyl-tRNA synthetases. Biochemistry. 2012;51(44):8705–29.
Bullwinkle TJ, Ibba M. Emergence and Evolution. In: Aminoacyl-tRNA Synthetases in Biology and Medicine edn. Edited by Kim S. Dordrecht: Springer Netherlands; 2014: 43–87.
Acknowledgements
We thank the patient and her family for their kind cooperation and Cipher Gene LCC for sequencing.
Funding
This work was supported by the National Nature and Science Foundation of China (No.82271509 & No.81771396), the Foundation of Jilin Provincial Key Laboratory of Pediatric Neurology (No.YDZJ202102CXJD021), the Project of Jilin Provincial Science and Technology Development Plan (No.YDZJ202201ZYTS676), the Natural Science Foundation of Jilin Province (No. YDZJ202201ZYTS090) and the Project of Jilin Medical and Health Talents (No.JLSWSRCZX2021053).
Author information
Authors and Affiliations
Contributions
Nuo Yang and Jianmin Liang contributed to the conception and design of the study. Nuo Yang, Limin Chen, Fan Yang, and Zuozhen Yang were in charge of the data analysis, and prepared Figs. 1, 2 and 3; Tables 1, and 2, supplementary Table 1. Nuo Yang and Yanfeng Zhang drafted the manuscript. Xuemei Wu, Yun Peng Hao, and Jianmin Liang commented on and revised the draft, and all authors have read and approved the final version of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Ethics Committee of Bethune First Hospital of Jilin University. Informed consent was obtained from all the participants and their legal guardian involved in the study.
Consent for publication
Written Informed consent from all the participants and patient's parents was obtained before conducting the WES, including the patient's clinical and imaging details in the manuscript for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table 1.
The candidate pathogenic genes variants in our patient.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, N., Chen, L., Zhang, Y. et al. Novel NARS2 variants in a patient with early-onset status epilepticus: case study and literature review. BMC Pediatr 24, 96 (2024). https://doi.org/10.1186/s12887-024-04553-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-024-04553-0