
Abstract
Background
Pediatric pulmonary hypertension (PH) is a serious and rare disease that is often derived from genetic mutations. Kabuki syndrome (KS) is a chromosomal abnormality disease that has its origin in the mutation of lysine methyltransferase 2D(KMT2D). Recent evidence has shown that KMT2D mutations are associated with pediatric pulmonary disorders. However, the relationship between the clinical courses of PH and the KMT2D mutation is reported in extremely few cases. Therefore, in this paper, a case was presented and previous literature was reviewed for better understanding of the correlation between pediatric PH and KMT2D mutations.
Case presentation
A 3-year-old girl was transferred to our center for severe cough, shortness of breath, fatigue and fever. Physical examination revealed facial deformities and growth retardation. Echocardiography showed a small atrial septal defect (ASD), and right heart catheterization indicated a significant increase in pulmonary vascular pressure and resistance. The genetic test suggested that she had a KMT2D gene mutation. The patient was finally diagnosed with KS. She was given targeted drugs to reduce pulmonary vascular pressure, but the effect was unsatisfactory.
Conclusions
KS can be complicated with multiple organ malformations and dysfunction. With the progress of next generation sequencing, an increasing number of new phenotypes related to KMT2D mutations have been reported. A bold hypothesis is proposed in this article, that is, PH may be a new phenotype associated with KMT2D mutations. It is suggested that KS and PH should be differentiated from each other to avoid delayed diagnosis and treatment in clinical practice. There is no specific drug for KS treatment. The prognosis of children with inherited PH is usually poor, and lung transplantation may increase their survival rates.
Similar content being viewed by others


Background
Pediatric pulmonary hypertension (PH) is a severe disease with significant morbidity and mortality. The causes of pediatric PH completely differ from those of adult PH [1]. The most frequently encountered types include idiopathic PH, PH due to congenital heart disease (CHD), and lung disease related PH. Kabuki syndrome (KS) is a chromosomal abnormality disease, which has different effects on the development and functions of multiple organ systems. However, there are few reports on pulmonary manifestations of KS patients, especially the correlation between KS and PH. Recent studies have revealed that interstitial lung disease is a new phenotype related to the KMT2D mutation of KS [2, 3]. Besides, KS patients have been reported to ultimately die of PH [4]. In this paper, a case of pediatric KS with a KMT2D mutation was presented. The patient had manifestations of growth retardation, an atrial septal defect (ASD), congenital hypothyroidism and severe PH. Moreover, it was proposed that PH might be a new phenotype of KMT2D mutations.
Case presentation
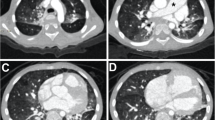
A 3-year-old girl was transferred to our center for severe cough, shortness of breath, fatigue and fever. Physical examination revealed facial deformities (Table 1) and growth retardation. The patient was diagnosed with hypothyroidism, CHD and PH by examinations at the age of 11 months old. Her transcutaneous finger oxygen saturation was 70%. Echocardiography showed a 0.9cm-wide ASD, a widened pulmonary artery, and severe tricuspid regurgitation (Fig. 1). Laboratory tests suggested that NT-proBNP was 3907pg/ml, the C-reaction protein level was 19.83mg/L, and the white blood cell was in the normal range, but the percentage of neutrophils increased to 78.7%, and (respiratory syncytial virus) RSV-IgM was positive. Liver function, kidney function, the level of autoimmune antibodies, and the erythrocyte sedimentation rate were normal. The patient was given anti-infection and cardiotonic drugs as well as respiratory support after admission.

Transthoracic echocardiogram. The four chamber view of cardiac ultrasound shows that the right heart was significantly enlarged,“*”indicates atrial septal defect, the shunt signal of blood flow from right to left can be seen at the defect, "↓" indicates moderate to severe tricuspid regurgitation. LV=left ventricular; LA=left atrial; RV=right ventricular; RA=right atrial
Meanwhile, the patient underwent right heart catheterization. The results indicated that the mean pulmonary arterial pressure (mPAP, 71mmHg) was markedly increased, the pulmonary vascular resistance (PVR, 27WU) also enlarged, and the pulmonary capillary wedge pressure (PCWP) is normal. Since the ASD was small and the slow flow rate could not result in the tremendous elevation in the pulmonary arterial pressure, the pathogenesis should be further investigated. No obvious abnormalities were found from the results of the pulmonary test, cardiac CTA and other routine etiological examinations in the patient. Nevertheless, the genetic inspection (the accession number for the whole-exome sequencing data is HRA005032) showed that the patient had a KMT2D (ENSMBL reference ID: NM_003482.3) exon 39 c.12209_12210del p.(Ser4070fs) mutation (Fig. 2). Based on the genetic results and abnormal countenance, the patient was diagnosed with KS type 1. The patient developed severe PH and poor oxygenation, and she was classified into the high-risk population by risk stratification was. Therefore, she received triple targeted pulmonary vascular pressure reduction treatment, i.e., Ambrisentan (2.5mg once daily) + Tadalafil (10mg once daily) + Remodulin (continuous subcutaneous pumping). The patient was followed up regularly after discharge, but the effect was unsatisfactory. The latest echocardiography reexamination showed no change in the size of the ASD, moderate tricuspid regurgitation, and estimated pulmonary arterial systolic pressure of 96mmHg.

Genetic testing results are as follows. A The result of chromosome karyotype analysis shows that: 46, XX [20],which is a normal karyotype. B The first generation sequencing verification result shows that the proposiatus had c. 12209_12210del heterozygous mutation on gene KMT2D, neither father(the second group) nor mother(the third group) had mutation, which was a new mutation
Discussion and conclusions
PH refers to changes in the pulmonary vascular structure or function caused by multiple etiologies and various pathogens. PH manifests clinical and pathophysiological syndromes due to pulmonary vascular resistance and elevated pulmonary vascular pressure. It can develop into right heart failure and even cause death. There are several factors leading to PH. Idiopathic PH, CHD-PH, and lung disease-related PH are more common in children than in adults. A PH patient can have two or more pathogenic factors simultaneously. To explore the underlying mechanism, the next-generation sequencing is conducted, which reveals genetic defects associated with pediatric PH.
KS is a chromosomal abnormality, which was firstly reported by Japanese scholars Niikawa [5] and Kuroki [6] in 1981. It can cause multiple anomalies, and postnatal growth retardation, hypotonia, and congenital organ malformations are its main clinical manifestations [7, 8]. KS is divided into KS type 1 and KS type 2. KS type 1 exhibits autosomal dominant inheritance [9], and it arises from the mutation of lysine specific methyltransferase 2D(KMT2D) (44%~76%) [10,11,12,13]. KS type 2 is originated from the mutation of lysine demethyltransferase 6A(KDM6A) located on the X chromosome (1%~6%), representing X-linked dominant inheritance [7, 14]. According to some previous literature, RAP1A and RAP1B gene mutations may be new and rare causes of KS [15]. Other studies have also shown that KDM6C, the homologue of KDM6A, is another H3K27 demethylase located on the Y chromosome, and it may act as a candidate gene for KS in male patients [16]. However, early diagnosis of KS remains a challenge in clinical practice since some typical phenotypes of KS only appear with age [17]. High-throughput sequencing technology is applicable to early and accurate diagnosis of KS at the genetic level.
At present, no research has proved a correlation between KS and PH, and the pulmonary phenotype of KS is also rarely discussed. Bang Tami J [3] analyzed CVID-related lung disorders in 2018, and he concluded that KS could cause interstitial lung disease. The conclusion was made based on the fact that KS patients shared similar immune disorders as variable immunodeficiency (CVID) patients. In 2019, Baldridge et al. [2] reported KS cases combined with interstitial lung disease, and they proposed that interstitial lung disease was a new phenotype associated with KMT2D variants. Moreover, in a case of KS with respiratory insufficiency, hematoxylin and eosin stained sections of the explanted lung demonstrated extensive alveolar remodeling and septal widening, many pulmonary fibrosis honeycomb structures, and prominent type II pneumocyte hyperplasia. The pathological findings suggested the possibility of PH. In addition, Baldridge also studied a KS patient with decreased physical strength. The echocardiogram of this patient showed elevated right ventricular systolic pressure and moderate tricuspid insufficiency. The electrocardiogram revealed right ventricular enlargement. Although the diagnosis of PH was not confirmed in this patient, the symptoms, echocardiogram and electrocardiogram results were suggestive of PH. In the study of Armstrong Linlea [4], a patient with KS died of PH. Therefore, a bold assumption is proposed in this paper that PH may be a new phenotype associated with KMT2D mutations.
There are two possible reasons for the PH in this patient as follows: (1) KMT2D gene mutation directly caused the PH, i.e., genetically associated PH(Class I), and for this hypothesis, the PH is considered as a new phenotype of KMT2D; (2) A pulmonary disease caused the PH, i.e., pulmonary disease-associated PH(Class III), for it has been reported that KMT2D gene mutation may result in pulmonary disease (usually interstitial lung disease). We are more inclined to agree with (1) for the following reasons: (1) Relevant examination for the patient showed no lung-related diseases like the interstitial lung disease or bronchial pulmonary hypoplasia, and currently, there is only PH, and so, it is reasonable to assume that the PH in this patient is genetically related, not associated with a lung disease caused by genetic mutation. (2) Usually, pulmonary disease-related PH is a moderate pulmonary hypertension with moderately elevated pressure and resistance, but the patient's high pulmonary arterial pressure and resistance are severe, which is not consistent with pulmonary disease-related PH. (3) At the same time, targeted drugs for lowering the pulmonary pressure control the progression of PH, which is consistent with the characteristics of Class I PH. Overall, we believe that the PH in this patient is a new phenotype of KMT2D mutation.
There is no specific drug for KS treatment. Children with inherited PH often have a poor prognosis. The drugs for preventing or reversing the progression of the disease have unsatisfactory effects. Lung transplantation may increase the survival rate of such patients.
Availability of data and materials
The raw sequencing data from this study have been deposited in the Genome Sequence Archive in BIG Data Center (https://bigd.big.ac.cn/), Beijing Institute of Genomics(BIG), Chinese Academy of Sciences, under the accession number: HRA005032.
Abbreviations
- PH:
-
Pulmonary hypertension
- KS:
-
Kabuki syndrome
- KMT2D :
-
Lysine methyltransferase 2D
- KDM6A :
-
Lysine demethyltransferase 6A
- CHD:
-
Congenital heart disease
- ASD:
-
Atrial septal defect
References
Rosenzweig EB, Abman SH, et al. Pediatric pulmonary arterial hypertension: updates on definition, classification, diagnostics and management. Eur Respir J. 2019;53(1):1801916.
Baldridge D, Spillmann Rebecca C, et al. Phenotypic expansion of KMT2D-related disorder: Beyond Kabuki syndrome. Am J Med Genet A. 2020;82:1053–65.
Bang Tami J, Richards J, et al. Pulmonary manifestations of common variable immunodeficiency. J Thorac Imaging. 2018;33:377–83.
Armstrong L, Aleck K, et al. Further delineation of Kabuki syndrome in 48 well-defined new individuals. Am J Med Genet A. 2005;null:265–72.
Niikawa N, Matsuura N, et al. Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. Pediatr. 1981;99:565–9.
Kuroki Y, Chyo H, et al. A new malformation syndrome of long palpebral fissures, large ears, depressed nasal tip, and skeletal anomalies associated with postnatal dwarfism and mental retardation. Pediatr. 1981;99:570–3.
Dentici ML, Di Pede A, et al. Kabuki syndrome clinical and molecular diagnosis in the first year of life. Arch Dis Child. 2015;100(2):158–64.
Courcet JB, Faivre L, et al. Clinical and molecular spectrum of renal malformations in Kabuki syndrome. J Pediatr. 2013;163(3):742–6.
Bogershausen N, Gatinois V, et al. Mutation Update for Kabuki Syndrome Genes KMT2D and KDM6A and Further Delineation of X-Linked Kabuki Syndrome Subtype 2. Hum Mutat. 2016;37(9):847–64.
Ng SB, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42(9):790–3.
Hannibal MC, Buckingham KJ, et al. Spectrum of MLL2 ALR mutations in 110 cases of Kabuki syndrome. Am J Med Genet. 2011;155A(7):1511–6.
Micale L, Fusco C, et al. Mutation spectrum of MLL2 in a cohort of Kabuki syndrome patients. Orphanet J Rare Dis. 2011;6:38.
Paulussen ADC, Stegmann APA, et al. MLL2 mutation spectrum in 45 patients with Kabuki syndrome. Hum Mutat. 2011;32(2):E2018-2025.
Paděrová J, Holubová A, et al. Molecular genetic analysis in 14 Czech Kabuki syndrome patients is confirming the utility of phenotypic scoring. Clin Genet. 2016;90(3):230–7.
Bogershausen N, Tsai IC, et al. RAP1-mediated MEK/ERK pathway defects in Kabuki syndrome. J Clin Invest. 2015;125(9):3585–99.
Walport LJ, Hopkinson RJ, et al. Human UTY(KDM6C) is a male-specific Nε-methyl Lysyl Demethylase. J Biol Chem. 2014;289(26):18302–13.
Vaux KK, Hudgins L, et al. Neonatal phenotype in Kabuki syndrome. Am J Med Genet. 2005;132A(3):244–7.
Acknowledgements
We would like to thank the patient’s family for their consent to publish this report. We also appreciated our medical team and their efforts to treat the patient.
Funding
The authors declare that they did not receive any source of funding for the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
XD, YL management of the patient, drafting the article, critical revision of the article; HZ, QS literature review, critical revision of the article; BJ, SL data collection; BJ, YL imaging evaluation. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed according to the Declaration of Helsinki. Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and accompanying images.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Deng, Xx., Jin, Bw., Li, Ss. et al. Pulmonary hypertension— a novel phenotypic hypothesis of Kabuki syndrome: a case report and literature review. BMC Pediatr 23, 429 (2023). https://doi.org/10.1186/s12887-023-04273-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-023-04273-x