
Abstract
Background
Congenital tibial hemimelia (CTH [MIM: 275220]) is a rare congenital limb deficiency that manifests as a shortened, curved, dysplastic or absent tibia with polydactyly. In previous studies, mutations of a distant sonic hedgehog (SHH) cis-regulator (ZRS) and a Shh repressor (GLI3) were identified.
Case presentation
Here, we admitted a 20-month-old boy who manifested with right tibial deformity, varus foot, ankle dislocation, and ipsilateral preaxial polydactyly. After genetic sequencing and data analysis, the results revealed a 443 A > G mutation in the father and a 536 C > T mutation in the mother in exon 2 of the Smoothed (SMO) gene at 7q32.1, with the coexistence of both mutant alleles in the proband/patient.
Conclusions
Our report suggests that even though not previously reported, SMO mutations may be associated with limb anomalies such as tibial hemimelia via Hh signaling in humans and has implications for genetic counseling.
Similar content being viewed by others


Background
Tibial hemimelia is a severe limb deformity with an extremely low incidence rate, with approximately one in one million (1/1000,000) live births [1, 2]. It mainly presents with a shortened, curved, dysplastic or absent tibia, a normal but usually proximal and/or distal dislocation of the fibula, varus ankle, and often polydactyly. According to previous reports, tibial hemimelia is associated with the zone of polarizing activity regulatory sequence (ZRS) and GLI3 mutations [3, 4].
Smoothed (SMO) gene, located at 7q32.1, is evolutionarily conserved among mammals and encodes a G protein-coupled receptor that interacts with the patched protein, a receptor for Hh proteins [5]. Previous studies have indicated that the SMO gene plays important roles in tumorigenesis via sonic hedgehog (Shh) signaling [6]. Here, we report a case manifested with right tibial hemimelia harboring the 443 A > G and 536 C > T mutations in SMO. To our knowledge, this is the first case report of tibial hemimelia associated with SMO mutation. We present the following case in accordance with the CARE reporting checklist.
Case presentation
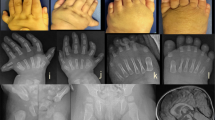
We admitted a 20-month-old boy with an abnormal right lower leg morphology since birth, accompanied by complex ipsilateral preaxial polydactyly. Consent for publication of the data was obtained from the family before the start of the study. There was no pain in the child’s right leg, normal growth in height and weight, and no abnormalities found in other parts of the body, such as the skin, hair, and face (Fig. 1). There were no abnormalities in the mother’s pregnancy and delivery and no other similar conditions in other family members. As he grew, the abnormalities of his right lower limb gradually worsened. At the time of his clinic visit, the patient had not yet learned to walk but was able to crawl normally. Physical examination revealed that the right tibia was anterolateral angulated and approximately 8.0 cm shorter than the left tibia. The right foot was internally rotated, with limited abduction and no dorsal extension at all. There was complex polydactyly on the lateral aspect of toe 1, and those toes could not move.

The patient’s right tibia was significantly anterolateral angulated, and approximately 8.0 cm shorter than the left, and the right foot was internal rotated. The photo was taken after the polydactylotomy
Radiological examination of the right leg revealed shortening, widening and thinning of the tibial stem, slender fibula, dislocation of the ankle, complex polydactyly of the right toes, and a redundant metatarsal forming an articulation with the superfluous toes (Fig. 2). A complete blood count, urine, blood biochemistry, chest radiograph, and ECG were within normal limits.

Radiological features of the proband. a X-ray showed polydactyly of the right toes, and a redundant metatarsal forming an articulation with the superfluous toes. b-f X-ray and computed tomography showed shortening, widening and thinning of the tibial stem, slender fibula, as well as dislocation of the and ankle
The child underwent polydactylotomy and right tibiofibular osteotomy reconstruction in our hospital. During the operation, we found that the morphology of the right tibia was changed from “tubular bone” to “flat bone”, and the medial margin of the tibia had a cartilage-like structure with localized thickened periosteum. The medullary cavity was not visible after cutting up the midshaft of the right tibia, but the bone hardness was basically normal. The prognosis of the child after surgery was favorable, and he is now able to stand on his own after 8 months of follow-up.
After searching the published reports, we considered this child’s case to be tibial hemimelia, which might be associated with genomic mutations. After obtaining informed consent (protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and approved by the Institutional Review Board and Ethical Committee at the West China Hospital of Sichuan University in China), we collected peripheral blood samples from the proband, his father and mother, extracted DNA, and performed next-generation whole exome sequencing (NGS) using the Illumina platform.
Data analysis and verification
NGS data were analyzed by Novogene (Tianjin, China). Briefly, sequencing results were aligned to the human genome (GRCh37/hg19) for SNP/INDEL calling: single nucleotide variants (SNVs) and indels were called with samtools to generate gVCF [7]. The copy number variants (CNVs) and structural variants (SV) were detected with Control-FREEC (v9.1) and LUMPY (v0.2.13) software, respectively. Then, annotation was performed using ANNOVAR (2017June8) [8]. Filtering of rare variants was performed as follows: (1) variants with an MAF less than 0.01 in 1000 genomic data (1000g_all) [9], esp6500siv2_all [10], gnomAD data (gnomAD_ALL and gnomAD_EAS) [11] and in the house Novo-Zhonghua exome database from Novogene; (2) only SNVs occurring in exons or splice sites (splicing junction 10 bp) were further analyzed since we are interested in amino acid changes. (3) Then, synonymous SNVs that were not relevant to the amino acid alternation predicted by dbscSNV were discarded. Small fragment nonframeshift (< 10 bp) indels in the repeat region defined by RepeatMasker were discarded. (4) Variations were screened according to scores of SIFT [12], Polyphen [13], MutationTaster [14] and CADD [15] software. The potentially deleterious variations are reserved if the score of more than half of these four software programs supports the harmfulness of variations [16]. Sites (> 2 bp) that did not affect alternative splicing were removed. To better predict the harmfulness of variation, the classification system of the American College of Medical Genetics and Genomics (ACMG) was used. The variations are classified into pathogenic, likely pathogenic, uncertain significance, likely benign and benign [17]. Then, variant-phenotype associations were analyzed with Phenolyzer and DisGetNet (V5.0). In our case, the SMO gene with two compound heterozygous variants (NM_005631: c.C536T:p.T179M and c.A443G:p.Q148R) was one of 25 genes in the candidate gene list found with Phenolyzer. Moreover, the SMO gene was the only gene found in DisGetNet associated with the phenotype of tibial hemimelia/polysyndactyly.
Based on the above analysis, we initially determined that the father and mother in this family each had a point mutation in exon 2 of the SMO gene at 7q32.1, and both missense mutations (father: SMO: NM_005631:c.A443G:p.Q148R or NC_000007.13:g. 128843336A > G; mother: SMO: NM_005631:c.C536T:p.T179M or NC_000007.13:g.128843429 C > T) and present in the child as compound heterozygous mutation, thus resulting in the disease (Fig. 3). Then, we designed primers (forward primer 5’-CTAGCAGGGCATCTGGAAGT-3’ and reverse primer 5’-TATACCCGGTCCTGCCCAAC − 3’) to amplify a 771 bp fragment covering these two mutation sites to validate the NGS result. Genomic DNA of peripheral blood from the father, mother, son (patient) and a wild type were applied to polymerase chain reaction, and amplified products were used for Sanger sequencing. The sequencing results showed a heterozygous 443 A > G mutation in the father, 536 C > T in the mother, and both in the proband but not in the normal control. This result verified the NGS findings (Fig. 4).

Pedigree of the family with of tibial hemimelia and the genetic sequence results

Mutation validation and protein structure modeling. a PCR sequencing result showing a heterozygous A443G mutation of SMO in the father and C536T in the mother, while compound heterozygous in the proband, but none in the wildtype (WT). b Both mutations are located in the CRD/Fz domain, which may potentially disrupt the protein/ligand or sterols binding affinity of SMO receptor. The black arrow indicates the mutation site and the red arrow indicates the presence of β-folding in WT but not in Q148R. the protein structures were simulated by pymol softwere. c Q148R substitution will change the local charge and polarity of the Helix structure, which was ploted with HeliQuest
Discussion and conclusion
The prevalence of limb anomalies in neonates is approximately 0.38% in China, with severe cases significantly affecting the survival and long-term quality of life of patients, most of whom require multiple operations [18, 19]. The malformations of tibial hemimelia may be unilateral or bilateral, and the tibia may be shortened, curved, dysplastic or even absent, along with various other limb deformities [20, 21]. There are different treatments for tibial hemimelia depending on the severity, and amputation is the most tried and proven method and is currently considered the gold standard of treatment for tibial hemimelia [20]. In our case, however, although the child’s shortened and “flattened” tibia had resulted in ankle dislocation and complete loss of standing ability, the parents could not accept amputation, and he ultimately underwent three operations for reconstructing the right lower limb structure and restoring function.
Tibial hemimelia is the result of abnormal limb bud development [22]. During embryonic development, bone formation is regulated by various signaling molecules, among which Hh is strongly involved. The Hh protein in mammals has three homologs, Shh, Indian hedgehog (Ihh) and Desert hedgehog (Dhh) [23]. At early stages of embryonic development, Shh regulates the formation of the zone of polarizing activity (ZPA) as a morphogen to establish the anterior-posterior (A-P) patterning of the embryonic limb bud [24, 25]. Ihh participates in the process of endochondral bone formation at late stages of embryonic development together with thyroid hormone-related peptide (PTHrP), which regulates growth plate and long bone development [26]. Previously reported tibial hemimelia associated with mutations in GLI3 and ZRS further validate the importance of the Shh signaling pathway in limb bud development [3, 4]. In addition to GLI3 and ZRS, Shh signaling is also regulated by the seven transmembrane protein SMO [27, 28].
SMO protein plays a key role in transducing Hh signaling. When the Hh ligand binds to the patched protein, the patched-mediated inhibition of SMO is released, allowing the signal to be transduced [29]. Although no limb abnormalities have been reported to be associated with SMO mutations, as mentioned above, it is speculated that SMO may also be involved in regulating limb development through the Hh signaling pathway. This case had two point mutations in exon 2 of the SMO gene at 7q32.1, and both were missense, which will cause a single amino acid substitution instead of a change in protein length. The SMO protein is an integral membrane receptor with an extracellular cysteine rich domain (CRD), also called the Fz domain, a transmembrane domain and an intracellular domain. Both of these mutations are located in the CRD/Fz domain (65-181Aa), which may potentially disrupt the protein/ligand or sterol binding affinity of the SMO receptor. For the maternally derived mutation (c.536 C > T;p.T179M;rs115491500) according to the ACMG criteria is classified as “likely pathogenic”; nevertheless, the paternal mutation (c.443 A > G;p.Q148R) is classified as of “uncertain significance”. However, Q148 is located in a Helix motif formed by 8 amino acids (144-RTLCQATR-151; PDB: 5V56), and the Q > R substitution will change the local charge and polarity, leading to confirmation change and structural disturbance (Fig. 4b and c). Moreover, this Q148R substitution may somehow affect disulfide bond formation between amino acids 147 and 156 C (147-CQATRGPCAIVERERGWPDFLRC-156; UniRule PROSITE-ProRule: PRU00090). Thus, we speculate that the structure or function of the SMO protein may have been disrupted in our case, affecting Hh signal transduction and leading to the malformation condition of the proband.
In conclusion, we report a case of tibial hemimelia that may be associated with point mutations in the SMO gene. The finding could help provide targeted gene sequencing technology to improve the pregnancy screening of this family and fetuses with similar presentations. The data also suggest that SMO mutations may also be involved in the development of limb abnormalities in humans, and further studies are needed to confirm this, potentially improving the process of genetic studies of limb development.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- SMO:
-
Smoothed
- Shh:
-
Sonic hedgehog
- ZRS:
-
Zone of polarizing activity regulatory sequence
- Ihh:
-
Indian hedgehog
- Dhh:
-
Desert hedgehog
- ZPA:
-
Zone of polarizing activity
- A-P:
-
Anterior-posterior
- PTHrP:
-
Thyroid hormone-related peptide
References
Weber M. Treatment of Tibial Hemimelia. 2015. p. 321–59.
Weber M, Schröder S, Berdel P, Niethard F. Register zur bundesweiten Erfassung angeborener Gliedmaßenfehlbildungen. Z Fur Orthopadie Und Ihre Grenzgebiete - Z ORTHOP GRENZGEB. 2005;143:534–8.
Norbnop P, Srichomthong C, Suphapeetiporn K. ZRS 406A > G mutation in patients with tibial hypoplasia, polydactyly and triphalangeal first fingers. J Hum Genet. 2014;59:467.
Deimling S, Sotiropoulos C, Lau K, Chaudhry S, Sturgeon K, Kelley S, et al. Tibial hemimelia associated with GLI3 truncation. J Hum Genet. 2016;61:443.
Moraes R, Zhang X, Harrington N, Fung J, Wu MF, Hilsenbeck S, et al. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development. 2007;134:1231–42.
Xie J, Murone M, Luoh SM, Ryan A, Qm G, Ch Z, et al. Activating smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–2.
Li H. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164.
Altshuler DM, Durbin RM, Abecasis G, Bentley DR, Chakravarti A, Clark AG, et al. A global reference for human genetic variation. Nature. 2015;526:68.
Tan R, Wang J, Wu X, Juan L, Zhang T, Ma R, et al. ERDS-Exome: a Hybrid Approach for Copy number variant detection from whole-exome sequencing data. IEEE/ACM Trans Comput Biol Bioinf. 2017;PP:1.
Holgersen EM, Gandhi S, Zhou Y, Kim J, Vaz B, Bogojeski J, et al. Transcriptome-wide Off-Target Effects of Steric-Blocking oligonucleotides. Nucleic Acid Ther. 2021;31(6):392–403.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4(7):1073–81.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7(8):575–6.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–5.
Muona M, Berkovic SF, Dibbens LM, Oliver KL, Maljevic S, Bayly MA, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet. 2015;47(1):39–46.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Shi Y, Zhang B, Kong F, Li X. Prenatal limb defects: epidemiologic characteristics and an epidemiologic analysis of risk factors. Medicine. 2018;97: e11471.
Thurmond A. Textbook of fetal abnormalities. Am J Roentgenol. 2001;176:622.
Paley D. Tibial hemimelia: new classification and reconstructive options. J Child Orthop. 2016;10:1–27.
Stevens C, Moore C. Tibial hemimelia in Langer–Giedion syndrome—possible gene location for tibial hemimelia. Am J Med Genet. 1999;85:409–12.
Weber M. Congenital leg deformities: tibial hemimelia. Congenital leg deformities: tibial hemimelia. 2006.
Echelard Y, Epstein D, St-Jacques B, Shen L, Mohler J, McMahon J, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1994;75:1417–30.
Anderson E, Peluso S, Lettice LA, Hill RE. Human limb abnormalities caused by disruption of hedgehog signaling. Trends Genet. 2012;28(8):364–73.
Hill RE, Heaney SJH, Lettice LA. Sonic hedgehog: restricted expression and limb dysmorphologies. J Anat. 2003;202(1):13–20.
Kronenberg H, Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6.
Price MA, Kalderon D. Proteolysis of the hedgehog signaling effector Cubitus interruptus requires phosphorylation by glycogen synthase kinase 3 and casein kinase 1. Cell. 2002;108(6):823–35.
Jia J, Amanai K, Wang G, Tang J, Wang B, Jiang J. Shaggy/GSK3 antagonizes hedgehog signalling by regulating Cubitus interruptus. Nature. 2002;416(6880):548–52.
Methot N, Basler K. Suppressor of fused opposes hedgehog signal transduction by impeding nuclear accumulation of the activator form of Cubitus interruptus. Development. 2000;127:4001–10.
Acknowledgements
We would like to thank Xianming Mo as a senior professor giving us constructive suggestions, Ph.D. Ran Lu and other members of the Laboratory of Stem Cell Biology, West China Hospital, Sichuan University for their generous help in methodology and reagents. We also thank Yun Yan for her generous help in protein structure analysis.
Funding
This research was supported by National Key R&D Program of China (2022YFC2703700, 2022YFC2703704), National Natural Science Foundation of China (31871449), Science and Technology Department of Sichuan (2022NSFSC1301 to Jing Chen; 2020YFS0108 to Xiaodong Yang), and 1·3·5 project for disciplines of excellence, West China Hospital, Sichuan University (ZYJC18003 to Yi Liu & 2021HXFH020 to Bo Xiang).
Author information
Authors and Affiliations
Contributions
XDY and SYP contributed equally to this work. SYP, XDY, BX and XYT carried out the study. XDY and SYP wrote the manuscript. JC supervised the project. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All procedures performed in studies involving human participants were conformed to the ethical guidelines of the 1975 Declaration of Helsinki and approved by the Institutional Review Board and Ethical Committee at the West China Hospital of Sichuan University in China. Consent for publication of the data was obtained from the family before the start of the study. The parents had gave their full, informed, written consent for their DNA to be sequenced, along with participation in any non-routine care procedures.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Competing interests
There is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, X., Pu, S., Xiang, B. et al. A novel smoothed (SMO) point mutation in congenital tibial hemimelia: a case report. BMC Pediatr 23, 424 (2023). https://doi.org/10.1186/s12887-023-04167-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-023-04167-y