
Abstract
Background
Periventricular nodular heterotopia (PNH), associated with FLNA mutations, is a rare clinical condition potentially associated with multiple systemic conditions, including cardiac, pulmonary, skeletal, and cutaneous diseases. However, due to a paucity of information in the literature, accurate prognostic advice cannot be provided to patients with the disease.
Case presentation
We report a 2-year-old female whose PNH was associated with a nonsense mutation in the q28 region of the X chromosome, in exon 31 of FLNA (c.5159dupA). The patient is currently seizure-free and has no congenital heart disease, lung disease or skeletal or joint issues, and her development is normal.
Conclusions
FLNA-associated PNH is a genetically-heterogeneous disease, and the FLNA mutation, c.5159dupA (p.Tyr1720*) is a newly identified pathogenic variant. FLNA characterization will help the clinical diagnosis and treatment of PNH and provide individualized genetic counseling for patients.
Similar content being viewed by others


Background
FLNA is located in the q28 region of the X chromosome [1, 2]. It encodes a widely expressed filamentous protein that acts on intracellular actin binding, and is involved in cell migration, mechano sensing, and cell signaling [3]. FLNA variants trigger X-linked filopathies which affect all organs, including the brain, bones, heart, and skin [4]. Periventricular nodular heterotopia (PNH) is strongly associated with FLNA mutations, which causes a loss of protein function, meaning developing neuronsfail to differentiate or migrate to the cortex in a timely manner [5]. This causes bilateral gray matter ectopia at the lateral ventricular rim, combined with a large occipital cisterna and hypoplasia of the cerebellum and corpus callosum [1, 6]. As FLNA is located on the X chromosome and is prevalent in females, males with PNH may die from severe complications prenatally or at an early age [6]. It is worth noting that the relevance of approximately 1/3 of all FLNA mutations is unknown, and that the clinical heterogeneity of FLNA mutations is extremely high [7, 8]. Therefore, no genotype-phenotype correlations have been identified [8], which is reflected by a paucity of literature on the subject.
We identified PNH in a 26-month-old girl admitted to our hospital for febrile seizures. The patient’s father had febrile seizures when he was a child, and the patient’s aunt had severe epilepsy and died in infancy, so we did MRI and genetic tests for this patient. Genetic tests identified FLNA mutations in exon 31, however, no mutational information in this sub-region was available from the literature. Currently, the girl is not experiencing any epilepsy and no developmental delays. We believe this case report and our literature review could provide individualized treatment plans and better prognoses for patients with FLNA-associated PNH disease.
Case introduction
The female patient was 26 months old. She was admitted to the Department of Pediatrics of the Affiliated Hospital of Hangzhou Normal University in November 2022 with a “2 hour fever and one seizure episode”. The child had one febrile seizure when she was 12 months old, and her aunt (mother’s sister) died of a severe neurological developmental abnormality at a young age. The father had one febrile seizure as a young child. Previously, the girl raised head at 3 months, sat at 8 months, talked at 12 months, walked at 13 months, and currently understood simple vocabulary with no obvious signs of developmental delay. Neurological physical examinations were negative. Post-admission ancillary examinations using photo-stimulated electroencephalogram and 24 h video EEG showed no abnormalities, and no epileptiform discharges were captured. Cardiac ultrasound morphology, structure, and hemodynamics did not show any significant abnormalities. A cranial magnetic resonance imaging (MRI) examination suggested gray matter heterotopia, corpus callosum dysplasia, and an arachnoid cyst in the occipital greater cisterna (Fig. 1).

Magnetic resonance imaging (MRI) of the patient suggests multiple nodular gray matter signal shadows in bilateral lateral ventricles, irregular morphology of bilateral lateral ventricles, and significant enlargement of the occipital greater cisterna. The arrow in figure a indicates a large occipital cisterna; The arrows in Figure b indicate multiple nodular gray matter signals in bilateral lateral ventricles; The arrow in figure c indicates hypoplasia of the cerebellum
The study was approved by the Medical Ethics Committee of Hangzhou Normal University Hospital (approval number 2021-(E2)-hs-059) in accordance with the ethical guidelines of the Declaration of Helsinki. Written informed consent was obtained from the patient’s guardian. We performed trio whole exon sequencing using targeted region capture high-throughput sequencing, and observed two variants in the patient’s FLNA gene. After QC was used to assess the sequencing quality of raw sequencing data and remove low-quality and joint-contaminated reads. The filtered data were sequenced with the human HG19 reference genome using BWA software (Burrows Wheeler Aligner) and the capture effect was assessed. GATK software was used to analyze single nucletide Variant (SNV) and Inde (INSERTION and deletion). 1000 Genomes (1000 Human Genome Dataset), Genome Aggregation Database dataset 2.1.1 and ExAC (The Exome) were used Aggregation Consortium dataset (Aggregation Consortium DATASET) screened the SNV and Indel obtained by analysis. The pathogenicity of false sense mutation and shear mutation was predicted using dbNSFP database. Reported mutations were screened using the Human Mendelian Genetic Database (OMIM), human Gene Mutation Database (HGMD) and Clinvar database. All mutation sites were classified using ACMG genetic variation classification criteria and guidelines. Finally, all possible pathogenic sites were verified by Sanger sequencing. One was in exon31,c.5159dupA:p.Tyr1720* (nucleotide duplication in coding region 5159, resulting in termination at tyrosine 1720) and was considered a heterozygous nonsense mutation. The other occurred at exon36, c.5764G > A:p.Val1922Met (mutation in nucleotide 5764 in the coding region (guanine to adenine), resulting in the mutation of valine (1922) to methionine), and was considered a heterozygous missense mutation (presently not significant). Genetic verification of the patient’s family history revealed a heterozygous mutation in c.5764G > A in the patient’s mother, and a hemizygous mutation in c.5764G > A in the patient’s (maternal) grandfather. Both the mother and (maternal) grandfather underwent cranial MRI, but no PNH was identified; both had no history of epilepsy and were currently healthy. When we processed this information, we hypothesized the child’s PNH was associated with the nonsense mutation in exon 31 of FLNA (c.5159dupA). FLNA mutations in the patient, the patient’s mother, and the patient’s (maternal) grandfather are shown (Figs. 2 and 3), and the family tree is shown (Fig. 4).

The patient (a) has a mutation in exon 31 of FLNA (red arrow); a heterozygous nonsense mutation in nucleotide duplication 5159 in the coding region, resulting in protein termination at tyrosine 1720. The patient’s mother and (maternal) grandfather have no mutation information in this region

Patient (a), patient’s mother (b) and patient’s (maternal) grandfather (c) have FLNA mutationsin exon36 (mutation sites are marked with red arrows). The patient has a heterozygous missense mutation in the coding region at nucleotide 5764 (guanine to adenine), resulting in mutation of amino acid 1922, valine to methionine. The patient’s mother has a heterozygous mutation in c.5764G > A, and the patient’s (maternal) grandfather has a hemizygous mutation in c.5764G > A

Patient’s family tree. Circle = female. Square = male. Black arrows = index patient. Black dots = FLNA with mutations. Slashes = death (this patient had a history of epilepsy). *completed FLNA testing in the family
Therefore, the patient had a heterozygous missense mutation in the coding region of FLNA at nucleotide 5764 (guanine to adenine), resulting in valine (1922) mutation to methionine. Her mother was heterozygous while her (maternal) grandfather was hemizygous for the mutation.
Literature review
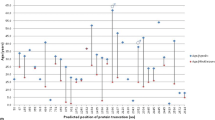
We searched PubMed for English-language studies published before February 20th, 2022, using “periventricular nodular heterotopias” AND “FLNA” OR “Grey matter heterotopias” AND “FLNA” terms. We sought studies on PNH associated with FLNA mutations, which provided mutation information, such as mutation type in exonic regions, descriptions of clinical symptoms, and images indicating cranial MRI alterations. We retrieved 19 publications covering diverse FLNA mutations in PNH patients (Fig. 5). Information on FLNA mutations, number of cases, patient gender, MRI imaging descriptions, clinical features, and levels of cognitive development were also gathered (Table 1).

The location of exonic FLNA mutations associated with periventricular nodular heterotopia. FLNA comprises 49 exons, including a new “poison” exon, 9N. Orange vertical bars represent exons, the purple horizontal line represents introns, numbers above orange bars are exon numbers, numbers in brackets (below orange bars) represent the number of cases included in mutation information, and different color dots represent different FLNA mutation types. This is a summary of literature search
Discussion
In previous PNH cases, patients were shown to have refractory epilepsy, cognitive and developmental impairment, and were mostly associated with a poor prognosis [5]. In our patient, we confirmed PNH was associated with FLNA, consistent with other FLNA-associated mutations in other patients with PNH and their family lines. However, with advanced precision medicine, diseases associated with FLNA mutations are now reported more frequently, with an increasing emphasis on genetic heterogeneity [6, 7, 27].
The diseases associated with FLNA mutation are known as X-linked filopathies due to the critical role of FLNA in organ development in humans [4]. PNH linked-FLNA mutations are associated with cardiovascular disease, malformations in the frontal face, congenital lung disease, excessive laxity of the skin and joints, and platelet abnormalities [28,29,30]. We observed a definite female prevalence for PNH associated-FLNA mutations, however, their overall prognosis was superior to males [31]. Moreover, in a larger number of cases, many patients were cognitively normal and had completed their university studies [32]. More interestingly, we showed that the proportion of febrile seizures was higher in patients with PNH [33, 34], consistent with our case who experienced these seizures and was subsequently diagnosed with PNH. However, associations between FLNA mutations and febrile seizures remain to be fully investigated.
From the literature, it was suggested that FLNA mutation type and exonic region could be correlated with clinical prognosis [13, 35, 36]. In males, survival and phenotype disease severity associated with missense mutations and distal truncation mutations were relatively positive, however, phenotypes associated with gene fragment insertions and deletions could be fatal [7]. From the literature, exon 2 mutations were the most reported; all were missense, with variable patient prognoses. Therefore, from the limited available information on FLNA-associated PNH, we hypothesize the mutation type and associated exonic region are not predictive of a clinical prognosis.
Our patient had no PNH family history, no current epilepsy, and cognitive and motor development was normal. Genetic characterization of the family showed the (maternal) grandfather and mother had a c.5764G > A mutation in exon 36 of FLNA, but MRIs showed no ectopic changes in their ventricular gray matter. For patients with a family history of febrile seizure and epilepsy, it is necessary to improve cranial MRI and genetic testing at the time of the first febrile seizure, which can lead to early diagnosis and prognosis. It was worth noting, in the literature, we observed no exon 36 FLNA mutation associations with PNH. When combined with our family’s genetic profile, we believe the missense mutation in this exon is not associated with PNH in our patient, but the c.5159dupA nonsense mutation in exon 31 may be the cause of her PNH. Importantly, this is the first PNH-associated case study in this exon in the literature. Based on patient clinical examinations, severe cardiac disease, pulmonary disease, and excessive skin and joint laxity have been ruled out, which suggests a good prognosis for this patient.
Conclusions
In clinical settings, PNH is a rare neuro developmental disease, therefore FLNA variants should be clarified as soon as possible after a PNH diagnosis. FLNA variants can cause X-linked filopathies, which potentially affect several important organs [32]. We reported a female child with PNH whose disease was associated with a nonsense mutation in exon31 of FLNA in the q28 region of the X chromosome. Currently, the patient is developing normally, with no seizures, and no congenital heart disease, lung disease, or skeletal and joint issues.
Diseases associated with FLNA variants are genetically heterogeneous, therefore, early and comprehensive clinical evaluations could help patient survival and social functioning in later life.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- PNH:
-
Periventricular nodular heterotopia
- FLNA :
-
Filamin A
References
Vriend I, Oegema R. Genetic causes underlying grey matter heterotopia. Eur J Paediatr Neurol. 2021;35:82–92.
Hiromoto Y, Azuma Y, Suzuki Y, et al. Hemizygous FLNA variant in West syndrome without periventricular nodular heterotopia. Hum Genome Var. 2020;7(1):43.
Jenkins ZA, Macharg A, Chang CY, et al. Differential regulation of two FLNA transcripts explains some of the phenotypic heterogeneity in the loss-of-function filaminopathies. Hum Mutat. 2018;39(1):103–13.
Wade EM, Halliday BJ, Jenkins ZA, O’Neill AC, Robertson SP. The X-linked filaminopathies: synergistic insights from clinical and molecular analysis. Hum Mutat. 2020;41(5):865–83.
Broix L, Jagline H, Ivanova E, et al. Mutations in the HECT domain of NEDD4L lead to AKT-mTOR pathway deregulation and cause periventricular nodular heterotopia. Nat Genet. 2016;48(11):1349–58.
Liu W, An D, Niu R, Gong Q, Zhou D. Integrity of the corpus callosum in patients with periventricular nodular heterotopia related epilepsy by FLNA mutation. Neuroimage Clin. 2018;17:109–14.
Parrini E, Ramazzotti A, Dobyns WB, et al. Periventricular heterotopia: phenotypic heterogeneity and correlation with Filamin A mutations. Brain. 2006;129(Pt 7):1892–906.
Meliota G, Vairo U, Ficarella R, Milella L, Faienza MF, D’Amato G. Cardiovascular, brain, and lung involvement in a newborn with a novel FLNA mutation: a case report and literature review. Adv Neonatal Care. 2022;22(2):125–31.
Fergelot P, Coupry I, Rooryck C, et al. Atypical male and female presentations of FLNA-related periventricular nodular heterotopia. Eur J Med Genet. 2012;55(5):313–8.
Parrini E, Mei D, Pisanti MA, et al. Familial periventricular nodular heterotopia, epilepsy and Melnick-Needles Syndrome caused by a single FLNA mutation with combined gain-of-function and loss-of-function effects. J Med Genet. 2015;52(6):405–12.
Zenker M, Rauch A, Winterpacht A, et al. A dual phenotype of periventricular nodular heterotopia and frontometaphyseal dysplasia in one patient caused by a single FLNA mutation leading to two functionally different aberrant transcripts. Am J Hum Genet. 2004;74(4):731–7.
Tsuneda SS, Torres FR, Montenegro MA, Guerreiro MM, Cendes F, Lopes-Cendes I. A new missense mutation found in the FLNA gene in a family with bilateral periventricular nodular heterotopia (BPNH) alters the splicing process. J Mol Neurosci. 2008;35(2):195–200.
van Kogelenberg M, Clark AR, Jenkins Z, et al. Diverse phenotypic consequences of mutations affecting the C-terminus of FLNA. J Mol Med (Berl). 2015;93(7):773–82.
Hehr U, Hehr A, Uyanik G, Phelan E, Winkler J, Reardon W. A filamin A splice mutation resulting in a syndrome of facial dysmorphism, periventricular nodular heterotopia, and severe constipation reminiscent of cerebro-fronto-facial syndrome. J Med Genet. 2006;43(6):541–4.
Oegema R, Hulst JM, Theuns-Valks SD, et al. Novel no-stop FLNA mutation causes multi-organ involvement in males. Am J Med Genet A. 2013;161A(9):2376–84.
LaPointe MM, Spriggs EL, Mhanni AA. Germline mosaicism in X-linked periventricular nodular heterotopia. BMC Neurol. 2014;14: 125.
Kasper BS, Kurzbuch K, Chang BS, et al. Paternal inheritance of classic X-linked bilateral periventricular nodular heterotopia. Am J Med Genet A. 2013;161A(6):1323–8.
Moro F, Carrozzo R, Veggiotti P, et al. Familial periventricular heterotopia: missense and distal truncating mutations of the FLN1 gene. Neurology. 2002;58(6):916–21.
de Wit MC, Tiddens HA, de Coo IF, Mancini GM. Lung disease in FLNA mutation: confirmatory report. Eur J Med Genet. 2011;54(3):299–300.
Lord A, Shapiro AJ, Saint-Martin C, Claveau M, Melançon S, Wintermark P. Filamin A mutation may be associated with diffuse lung disease mimicking bronchopulmonary dysplasia in premature newborns. Respir Care. 2014;59(11):e171-7.
Hommel AL, Jewett T, Mortenson M, Caress JB. Juvenile muscular atrophy of the distal upper extremities associated with x-linked periventricular heterotopia with features of Ehlers-Danlos syndrome. Muscle Nerve. 2016;54(4):794–7.
Reinstein E, Chang BS, Robertson SP, Rimoin DL, Katzir T. Filamin A mutation associated with normal reading skills and dyslexia in a family with periventricular heterotopia. Am J Med Genet A. 2012;158A(8):1897–901.
Sheen VL, Jansen A, Chen MH, et al. Filamin A mutations cause periventricular heterotopia with Ehlers-Danlos syndrome. Neurology. 2005;64(2):254–62.
Guerrini R, Mei D, Sisodiya S, et al. Germline and mosaic mutations of FLN1 in men with periventricular heterotopia. Neurology. 2004;63(1):51–6.
Parrini E, Mei D, Wright M, Dorn T, Guerrini R. Mosaic mutations of the FLN1 gene cause a mild phenotype in patients with periventricular heterotopia. Neurogenetics. 2004;5(3):191–6.
de Wit MC, de Coo IF, Lequin MH, Halley DJ, Roos-Hesselink JW, Mancini GM. Combined cardiological and neurological abnormalities due to filamin A gene mutation. Clin Res Cardiol. 2011;100(1):45–50.
Guglielmi V, Floris R, D’Adamo M, Garaci F, Novelli G, Sbraccia P. Massive obesity and hyperphagia in posterior bilateral periventricular heterotopias: case report. BMC Med Genet. 2016;17:18.
Kinane TB, Lin AE, Lahoud-Rahme M, Westra SJ, Mark EJ. Case 4-2017. A 2-Month-Old girl with growth retardation and respiratory failure. N Engl J Med. 2017;376(6):562–74.
Mercer CL, Andreoletti G, Carroll A, Salmon AP, Temple IK, Ennis S. Familial Ebstein Anomaly: whole exome sequencing identifies novel phenotype Associated with FLNA. Circ Cardiovasc Genet. 2017;10(6):e001683.
Riccio MP, D’Andrea G, Sarnataro E, Marino M, Bravaccio C, Albert U. Bipolar disorder with Melnick-Needles syndrome and periventricular nodular heterotopia: two case reports and a review of the literature. J Med Case Rep. 2021;15(1):495.
El Chehadeh S, Faivre L, Mosca-Boidron AL, et al. Large national series of patients with Xq28 duplication involving MECP2: delineation of brain MRI abnormalities in 30 affected patients. Am J Med Genet A. 2016;170A(1):116–29.
Lange M, Kasper B, Bohring A, et al. 47 patients with FLNA associated periventricular nodular heterotopia. Orphanet J Rare Dis. 2015;10:134.
Arya R, Spaeth C, Zhang W. Epilepsy phenotypes associated with MAP1B-related brain malformations. Epileptic Disord. 2021;23(2):392–6.
Fallil Z, Pardoe H, Bachman R, et al. Phenotypic and imaging features of FLNA-negative patients with bilateral periventricular nodular heterotopia and epilepsy. Epilepsy Behav. 2015;51:321–7.
Moutton S, Fergelot P, Naudion S, et al. Otopalatodigital spectrum disorders: refinement of the phenotypic and mutational spectrum. J Hum Genet. 2016;61(8):693–9.
Dissanayake R, Senanayake MP, Fernando J, Robertson SP, Dissanayake V, Sirisena ND. Frontometaphyseal dysplasia 1 in a patient from Sri Lanka. Am J Med Genet A. 2021;185(4):1317–20.
Acknowledgements
Not applicable.
Funding
This study was supported by the Ministry of Science and Technology of China (2021ZD0201705) and Zhejiang Medical Health Science and Technology Project (2023KY187).
Author information
Authors and Affiliations
Contributions
L Y: Conceptualization, methodology, formal analysis, resources, writing—original draft preparation; G Sh W: formal analysis, writing—original draft preparation; H M Y: data curation; M L P: data curation; Y F Zh: project administration, manuscript revision proofreading. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The experimental protocol was established, according to the ethical guidelines of the Helsinki Declaration and was approved by the Medical Ethics Committee of The Affiliated Hospital of Hangzhou Normal University, approval number: 2021-(E2)-HS-059. Written informed consent was obtained from individual or guardian participants.
Consent for publication
All children have obtained the informed consent of their guardians.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yang, L., Wu, G., Yin, H. et al. Periventricular nodular heterotopias is associated with mutation at the FLNA locus-a case history and a literature review. BMC Pediatr 23, 346 (2023). https://doi.org/10.1186/s12887-023-04161-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-023-04161-4