
Abstract
Background
Multiple sulfatase deficiency (MSD) is a rare lysosomal storage disorder caused due to pathogenic variants in the SUMF1 gene. The SUMF1 gene encodes for formylglycine generating enzyme (FGE) that is involved in the catalytic activation of the family of sulfatases. The affected patients present with a wide spectrum of clinical features including multi-organ involvement. To date, almost 140 cases of MSD have been reported worldwide, with only four cases reported from India. The present study describes two cases of late infantile form of MSD from India and the identification of a novel missense variant in the SUMF1 gene.
Case presentation
In case 1, a male child presented to us at the age of 6 years. The remarkable presenting features included ichthyosis, presence of irritability, poor social response, thinning of corpus callosum on MRI and, speech regression. Clinical suspicion of MSD was confirmed by enzyme analysis of two sulfatase enzymes followed by gene sequencing. We identified a novel missense variant c.860A > T (p.Asn287Ile) in exon 7 of the SUMF1 gene. In case 2, a two and a half years male child presented with ichthyosis, leukodystrophy and facial dysmorphism. We performed an enzyme assay for two sulfatases, which showed significantly reduced activities thereby confirming MSD diagnosis.
Conclusion
Overall, present study has added to the existing data on MSD from India. Based on the computational analysis, the novel variant c.860A > T identified in this study is likely to be associated with a milder phenotype and prolonged survival.
Similar content being viewed by others


Background
Multiple sulfatase deficiency (MSD) (OMIM#272200) is a rare lysosomal storage disorder with an incidence of 1 in 1.4 million newborns [1]. It occurs due to a defect in the SUMF1 gene, located on chromosome 3p26.1, that encodes for formylglycine generating enzyme (FGE) [2,3,4]. FGE is involved in the posttranslational modification and catalytic activation of the family of sulfatase enzymes. The FGE protein catalyzes a cysteine residue shared by all sulfatase enzymes to form formylglycine. This step is critical for their enzymatic functions. As a result, any defect in the FGE can lead to deficiency in the sulfatase enzyme family along with increased levels of sulfated lipids and mucopolysaccharides inside the cells [5]. In humans, there are seventeen types of sulfatases of which nine are associated with MSD [6]. The FGE protein catalyzes a cysteine residue shared by all sulfatase enzymes to form formylglycine. This step is critical for their enzymatic functions. As a result, any defect in the FGE can lead to deficiency in the sulfatase enzyme family along with increased levels of sulfated lipids and mucopolysaccharides inside the cells [5].
MSD patients present with a variable clinical spectrum and are influenced by the severity of FGE protein instability and residual catalytic ability. The three types of MSD based on the clinical presentation have been described, that include, neonatal, late infantile, and juvenile. Of these, neonatal is the most severe form whereby affected individuals are presented with coarse facial features, skeletal abnormalities, neurologic deterioration, ichthyosis, and mental retardation [7]. Late infantile is the most common form of MSD and is characterized by normal development in early childhood and gradual regression of psychomotor skills during late childhood [7].
Overall, approximately 143 cases of MSD have been reported worldwide [8] with very few cases of MSD from India [9,10,11]. In the present study, we describe two cases of MSD from India with a novel SUMF1 variant identified in one case along with the review of literature.
Case presentation
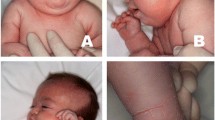
Case 1: A male child born to a second-degree consanguineous Muslim couple from Ahmedabad, Gujarat presented at our clinic at 6 years of age (Fig. 1A).

Case 1. A: Pedigree chart, proband is shown by arrow, B: Clinical photograph of the proband showing no facial dysmorphism, C: MRI of the proband showing mild thinning of posterior aspect of corpus callosum, D: Back region of the proband showing ichthyosis
The index case was delivered at full term with a birth weight of 2250 g and had delayed cry. At the age of 9 months, he was hospitalized due to diarrhoea. The child contracted Tuberculosis (TB) at 4 years of age and received a TB regimen therapy for 9 months. He showed regression of speech at 2.5 years of age. He had a poor response to social interactions and had aggressive behaviour. At first, the child was referred to a paediatric neurologist who suspected a case of syndromic autism spectrum disorder. His sleep EEG study showed intractable epilepsy activity from the left occipital hemisphere with spread to the opposite hemisphere and diffuse damage from the right hemisphere. In addition, his MRI showed mild thinning of the posterior aspect of the body of the corpus callosum with no evidence of cerebral and cerebellar atrophy (Fig. 1B). Subsequently, the child was referred to our centre to carry out additional investigations. On examination, the remarkable observation noted was the presence of ichthyosis on both the legs, back, and abdomen (Fig. 1C). There was no facial dysmorphism observed. He could not speak and was restless and babbling. Primary investigations of karyotype and Fragile-X were normal in the proband. Considering abnormal MRI and the clinical indications, we suspected metachromatic leukodystrophy (MLD) or MSD. Hence, investigation for lysosomal enzymes arylsulfatase-A, arylsulfatase-B, and beta-galactosidase was carried out from leukocytes using 4-Mu fluorogenic synthetic substrates. Leukocytes were separated from the whole blood of the patient and all the enzyme assays were analyzed using spectrophotometric and fluorometric methods as described earlier [12, 13]. We found low level of arylsulfatase-A (0.08 nmol/hr/mg protein) (NR: 0.6- 4.99 nmol/hr/mg protein) and arylsulfatase-B (0.23 nmol/hr/mg protein) enzyme activities (NR: 0.61- 9.6 nmol/hr/mg protein) with normal activity of the beta-galactosidase enzyme. Considering low activity of both sulfatases: arylsulfatase-A and arylsulfatase-B, MSD was the likely diagnosis in the proband. Furthermore, for molecular confirmation, we performed SUMF1 gene-sequencing study to determine the underlying causative variant. Genomic DNA was extracted from the peripheral blood of the proband using the desalting protocol [14]. Sanger sequencing of all the exons and exon–intron boundaries of the SUMF1 gene (NM_182760.4) in the proband detected a homozygous missense variant c.860A > T (p.Asn287Ile) in exon 7. The variant c.860A > T has not been reported in the 1000 genomes and gnomAD databases. Also, in-silico predictions of the variant using Polyphen-2 and SIFT were found to be damaging. The variant was classified as likely pathogenic as per the ACMG-AMP guidelines and ClinGen framework [15,16,17] with the following criteria – PM1 (supporting), PM2 (moderate), PP3 (supporting) and, PS3 (moderate). Following this, a parental study was carried out that confirmed them to be heterozygous carriers for the same variant in the SUMF1 gene (Fig. 2).

A, B, C: Sanger sequencing chromatogram of the proband and his parents
Case 2: A two and half-year-old male child, born to a non-consanguineous Muslim couple was referred to our clinic. The notable features included ichthyosis and facial dysmorphism. His MRI study showed leukodystrophy, which is a classical observation seen in patients with MLD and Krabbe disease. Based on these observations, child was investigated for lysosomal enzyme activity from leukocytes for arylsulfatase-A. However, due to presence of ichthyosis, MSD was also suspected and hence activity of another sulfatase: N-acetyl-galactosamine-6-sulfate-sulfatase was assessed as per the method earlier described [18]. The proband showed significantly low activity of arylsulfatase-A enzyme (0.045 nmol/hr/mg protein) and N-acetyl-galactosamine-6-sulfate-sulfatase enzyme (0.041 nmol/hr/mg protein) (NR: 2.8- 21.6 nmol/hr/mg protein) with the normal activity of beta-galactosidase enzyme. This confirmed the diagnosis of MSD in the proband. However, at the time of the investigation, the patient’s family refused to carry out further molecular study and hence the causative variant was not identified. Furthermore, the family was lost to follow-up and hence there is a lack of clinical, MRI pictures and details regarding the progression of the disease condition in order to comment on the severity.
Discussion and conclusion
MSD is an ultra-rare lysosomal storage disorder characterized by a deficiency of the FGE protein. The key presenting features of MSD are neurological complications, developmental delay, skeletal and dermatological abnormalities [8]. Of these, ichthyosis is the most frequently reported sign (~ 71% cases) followed by organomegaly (57% cases) and dysostosis multiplex (56% cases). Ichthyosis was present in both our cases, which is consistent with the earlier observation [8]. Facial dysmorphism, which is another common sign in MSD patients, was seen in only one of the two cases. Strikingly, skeletal abnormalities and organomegaly, which are generally common in MSD patients, were absent in both cases. Hijazi et al. also made a similar observation where none of the patients showed organomegaly and only 2/6 patients had a skeletal abnormality. This explains the wide clinical variability observed in MSD cases. The common MRI findings in MSD patients include leukodystrophy, cerebral and cerebellar atrophy, and hydrocephalus [19]. Interestingly, both our patients showed abnormal MRI with thinning of the corpus callosum in one case and leukodystrophy in another. For the first time, Hijazi et al. recorded autistic features in MSD patients [19]. We noted a similar finding in the proband from case 1. This suggests that autism spectrum disorder is likely to be a secondary manifestation arising due to MSD and often misdiagnosed as autism.
MSD is classified into three types depending on the age of onset and severity. Schlotawa and his group analysed the correlation between the onset of key signs and the survival age of the patient [8]. They showed neurological and skeletal signs before 24 months of age to be negatively associated with survival. As opposed to this, the presence of only ichthyosis after 36 months correlated with longer survival. Both of our patients are likely to have longer survival as both are alive to date. Yet, it is critical to note that cardiovascular and respiratory involvement is associated with reduced survival and is independent of onset age. Also, respiratory complications have been the most common cause of death in the case of MSD patients [8]. Though, none of our patients have shown respiratory complications yet. A recent study by Beck-Wödl et al. compared the clinical course of two patients with MSD to a broader cohort of MLD patients. Their observations suggested that patients with MSD show early onset of motor symptoms as compared to patients with juvenile MLD, but the disease progression is slow as compared to the juvenile MLD patients [20]. This is similar to what we observed for proband in case 1. Thus, this can be a key clinical observation in differentiating MSD patients from MLD patients.
Detecting elevated levels of glycosaminoglycan’s and other sulphated sugars in the patient’s urine sample can aid in the diagnosis of MSD and also to rule out arylsulfatase-A pseudo deficiency which is a common observation in ~ 7–12% of healthy population [21]. However, in the present study, these investigations were not carried out, as both cases had a strong clinical suspicion and positive lysosomal enzyme study. Based on this, MSD was considered as the most likely differential diagnosis.
The primary diagnosis of MSD can be carried out by lysosomal enzyme study followed by confirmative genetic investigation. It is important to note that the method of diagnosis does not affect patient survival in MSD; hence, either of the options can be used for the purpose of diagnosis [8]. The key advantage in cases where a molecular diagnosis has been made is that prenatal testing can be offered for future pregnancies. Biochemical diagnosis is achieved by reduced activity of two or more sulfatases whereas molecular testing determines the causative variant in the SUMF1 gene. At the molecular level, the clinical heterogeneity is explained by the amount of residual enzyme activity and the degree of protein stability [22]. In proband of case 1, we have identified a novel missense variant c.860A > T in the SUMF1 gene. So far, a majority of missense mutations have been reported, followed by nonsense and splicing mutations majority in the C-terminal domain of the FGE protein [22]. As the variant identified in our case is novel, we have carried out a computational analysis to determine its pathogenicity. We performed a structural study of the mutant SUMF1 using the crystallographic structure of FGE (PDB ID: 1Y1E) as the template. The native and mutant structure was modelled using a web server Swiss Model of Expasy. The two models were superimposed and aligned using the tool UCSF Chimera to understand the effect of the variant [23]. The superimposed model suggests that there is no significant conformational change in the loop region due to the presence of mutant amino acids Ile at position 287 of the protein (Supplementary Fig. 1). However, due to the change of a hydrophilic amino acid asparagine to a hydrophobic amino acid isoleucine, there is likely to be a disruption in protein binding resulting in overall instability of the protein.
High percentage of MSD cases have been reported from the USA, Saudi Arabia and, Turkey with thirty-one, twenty-six and, fifteen cases respectively. While from India only four cases have been reported with molecular study in one of them showing compound heterozygous missense mutation in exon 3 and insertion mutation in exon 5 as shown in Table 1. The majority of mutations have been identified in exon 5, 6 and, 9 suggesting them as the hot spot region. However, the present study has identified a missense mutation in exon 7 suggesting the possibility of ethnographic variability. Previously, a couple of studies have analyzed the functional effects of a subset of SUMF1 variants on the FGE protein [24,25,26] with the correlation between genotype and phenotype in MSD depending on protein stability and residual activity of mutant FGE protein. However, as more novel variants are being identified in the SUMF1 gene, functional and computational studies to predict the consequence of these variants at the molecular and clinical level need to be performed. The milder phenotype in case one in the present study could be attributed to the missense mutation affecting only protein stability. Thus, present study will add to the existing understanding of genotype–phenotype correlation in MSD and can aid in predicting disease severity and providing genetic counselling to the affected families.
There is a high degree of clinical variability seen in MSD patients, with ichthyosis and developmental delay being the common signs. At present, there is no treatment available, however, the involvement of a multidisciplinary team can aid in the holistic management of MSD patients. It is evident from the data in the literature that exon 6 of the SUMF1 gene is the hotspot for mutation; hence, this exon can be preliminary screened in MSD patients to identify the causative variant. Lastly, the variant identified in the present study, c.860A > T, based on the computational analysis is likely to have a relatively milder phenotype and prolonged survival.
Availability of data and materials
Not applicable.
Abbreviations
- MSD:
-
Multiple sulfatase deficiency
- SUMF1:
-
Sulfatase Modifying Factor1
- FGE:
-
Formyl glycine generating enzyme
- TB:
-
Tuberculosis
- MRI:
-
Magnetic resonance imaging
- EEG:
-
Electroencephalography
- DNA:
-
Deoxyribonucleic acid
- PDB:
-
Protein data bank
References
Eto Y, Ito T, Kobayashi H. [Multiple sulfatase deficiency]. Ryoikibetsu Shokogun Shirizu. 1998;19 Pt 2:385–8.
Adang LA, Schlotawa L, Groeschel S, Kehrer C, Harzer K, Staretz-Chacham O, et al. Natural history of multiple sulfatase deficiency: Retrospective phenotyping and functional variant analysis to characterize an ultra-rare disease. J Inherit Metab Dis. 2020;43(6):1298–309.
Garavelli L, Santoro L, Iori A, Gargano G, Braibanti S, Pedori S, et al. Multiple sulfatase deficiency with neonatal manifestation. Ital J Pediatr. 2014;17(40):86.
Ahrens-Nicklas R, Schlotawa L, Ballabio A, Brunetti-Pierri N, De Castro M, Dierks T, et al. Complex care of individuals with multiple sulfatase deficiency: Clinical cases and consensus statement. Mol Genet Metab. 2018;123(3):337–46.
Hirst L, Abou-Ameira G, Uudelepp ML. Multiple Sulfatase Deficiency (MSD): review of the literature and case reports of two siblings with dental caries and trauma. Case Rep Pediatr. 2021;2021:6611548.
Appel MJ, Bertozzi CR. Formylglycine, a post-translationally generated residue with unique catalytic capabilities and biotechnology applications. ACS Chem Biol. 2015;10(1):72–84.
Schlotawa L, Adang LA, Radhakrishnan K, Ahrens-Nicklas RC. Multiple sulfatase deficiency: a disease comprising mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. Int J Mol Sci. 2020;21(10):3448.
Schlotawa L, Preiskorn J, Ahrens-Nicklas R, Schiller S, Adang LA, Gärtner J, et al. A systematic review and meta-analysis of published cases reveals the natural disease history in multiple sulfatase deficiency. J Inherit Metab Dis. 2020;43(6):1288–97.
Kotecha UH, Movva S, Sharma D, Verma J, Puri RD, Verma IC. Molecular evaluation of a novel missense mutation & an insertional truncating mutation in SUMF1 gene. Indian J Med Res. 2014;140(1):55–9.
Bharucha BA, Naik G, Savliwala AS, Joshi RM, Kumta NB. Siblings with the Austin variant of metachromatic leukodystrophy multiple sulfatidosis. Indian J Pediatr. 1984;51(411):477–80.
Nalini A, Christopher R. Cerebral glycolipidoses: clinical characteristics of 41 pediatric patients. J Child Neurol. 2004;19(6):447–52.
Baum H, Dodgson KS, Spencer B. The assay of arylsulphatases A and B in human urine. Clin Chim Acta Int J Clin Chem. 1959;4(3):453–5.
Suzuki K. [61] Enzymatic diagnosis of sphingolipidoses. In: Methods in Enzymology. Academic Press; 1987. p. 727–62. (Complex Carbohydrates Part E; vol. 138). Available from: https://www.sciencedirect.com/science/article/pii/0076687987380632. [Cited 2023 Feb 8]
Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med Off J Am Coll Med Genet. 2015;17(5):405–24.
Biesecker LG, Harrison SM, ClinGen Sequence Variant Interpretation Working Group. The ACMG/AMP reputable source criteria for the interpretation of sequence variants. Genet Med Off J Am Coll Med Genet. 2018;20(12):1687–8.
Zhang J, Yao Y, He H, Shen J. Clinical Interpretation of Sequence Variants. Curr Protoc Hum Genet. 2020;106(1):e98.
van Diggelen OP, Zhao H, Kleijer WJ, Janse HC, Poorthuis BJ, van Pelt J, et al. A fluorimetric enzyme assay for the diagnosis of Morquio disease type A (MPS IV A). Clin Chim Acta Int J Clin Chem. 1990;187(2):131–9.
Hijazi L, Kashgari A, Alfadhel M. Multiple Sulfatase Deficiency: A Case Series With a Novel Mutation. J Child Neurol. 2018;33(13):820–4.
Beck-Wödl S, Kehrer C, Harzer K, Haack TB, Bürger F, Haas D, et al. Long-term disease course of two patients with multiple sulfatase deficiency differs from metachromatic leukodystrophy in a broad cohort. JIMD Rep. 2021;58(1):80–8.
Herz B, Bach G. Arylsulfatase A in pseudodeficiency. Hum Genet. 1984;66(2–3):147–50.
Schlotawa L, Ennemann EC, Radhakrishnan K, Schmidt B, Chakrapani A, Christen HJ, et al. SUMF1 mutations affecting stability and activity of formylglycine generating enzyme predict clinical outcome in multiple sulfatase deficiency. Eur J Hum Genet EJHG. 2011;19(3):253–61.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–12.
Annunziata I, Bouchè V, Lombardi A, Settembre C, Ballabio A. Multiple sulfatase deficiency is due to hypomorphic mutations of the SUMF1 gene. Hum Mutat. 2007;28(9):928.
Cosma MP, Pepe S, Parenti G, Settembre C, Annunziata I, Wade-Martins R, et al. Molecular and functional analysis of SUMF1 mutations in multiple sulfatase deficiency. Hum Mutat. 2004;23(6):576–81.
Schlotawa L, Steinfeld R, von Figura K, Dierks T, Gärtner J. Molecular analysis of SUMF1 mutations: stability and residual activity of mutant formylglycine-generating enzyme determine disease severity in multiple sulfatase deficiency. Hum Mutat. 2008;29(1):205.
Schlotawa L, Radhakrishnan K, Baumgartner M, Schmid R, Schmidt B, Dierks T, Gärtner J. Rapid degradation of an active formylglycine generating enzyme variant leads to a late infantile severe form of multiple sulfatase deficiency. Eur J Hum Genet. 2013;21(9):1020–3. https://doi.org/10.1038/ejhg.2012.291. Epub 2013 Jan 16. PMID: 23321616; PMCID: PMC3746267.
Acknowledgements
We are grateful to the family of the patients for their kind co-operation and permission. Also thankful to Mrs Riddhi Tikhe for biochemical enzyme study.
Funding
Partial funding for the said work has been received from Gujarat State Biotech mission (GSBTM), Govt of Gujarat, Dept of Science and Technology (Grant No. GSBTM/JDR &D/608/2020/456–458). The funding agency was not involved in the study design, specimen collection, analysis, interpretation and preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed experiments: JJS, HS and FJS. Patient recruitment and clinical analysis: DJ, SS, CD, FS and JJS. Sequencing, data analysis and interpretation: JS and HS. Write first draft of the manuscript: KB, PR, AN, JJS. Made critical revisions and approved final version: FS and JJS. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The present case under submission has been approved by the institutional ethics committee (FRIGE/IEC/18/2020) and was in accordance with the Helsinki declaration. Written informed consent was obtained from the parents before enrolling for the investigations [This was in accordance with the requirement of the institutional ethics committee].
Consent for publication
Written informed consent was obtained from parents for publication of identifying images and clinical details since the patient was under the age of 18 years.
Competing interests
The authors declare that they have no conflicts of interests (financial or non-financial).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure 1.
Superimposed native structure (blue) and mutant structure (brown) of the FGE protein produced using UCSF chimera. The purple highlighted portion is the wild-type residue and the blue highlighted portion is the mutant residue.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Sheth, J., Shah, S., Datar, C. et al. Late infantile form of multiple sulfatase deficiency with a novel missense variant in the SUMF1 gene: case report and review. BMC Pediatr 23, 133 (2023). https://doi.org/10.1186/s12887-023-03955-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-023-03955-w