
Abstract
Background
Idiopathic or transient neonatal cholestasis (TNC) represents a group of cholestatic disorders with unidentified origin and remains a diagnosis of exclusion. Dysfunction of hepatobiliary transporters mediating excretion of biliary constituents from hepatocytes may play a central role in the pathogenesis of cholestasis. Despite variants of bile salt (BS) export pump (BSEP/ABCB11) have already been described in TNC, the pathogenic role of BSEP dysfunction in TNC remained so far elusive.
Case presentation
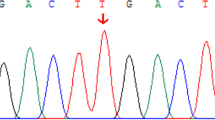
We report on a newly-identified heterozygous ABCB11 missense variant (c.1345G > A, p.Glu449Lys) which was associated with prolonged cholestasis in a term infant after a complicated neonatal period. Moreover, we show for the first time almost completely abolished BSEP expression on the hepatocellular membrane in TNC.
Conclusion
This report demonstrates for the first time a close association between the prolonged cholestasis in infancy and impaired BSEP expression on the hepatocyte canalicular membrane in a heterozygous carrier of newly-identified ABCB11 variant.
Similar content being viewed by others


Background
Idiopathic or transient neonatal cholestasis (TNC) represents a group of intrahepatic cholestatic disorders in infancy with poorly understood pathogenesis. It is characterized by onset of cholestasis in the first week of life, elevation of biochemical parameters of liver injury and cholestasis for several months and their normalization usually by the age of 1 year [1]. Interestingly, despite the complicated neonatal period (asphyxia, sepsis, parenteral nutrition, etc.) is associated with a higher incidence of TNC [1], only some of the infants admitted to the neonatal intensive care unit develop TNC, suggesting involvement of genetic predisposing mechanisms.
Bile salt (BS) excretion from hepatocytes is mediated by BS-specific canalicular transporter the bile salt export pump (BSEP/ABCB11). Mutations leading to complete loss of BSEP function cause progressive familial intrahepatic cholestasis type 2 (PFIC 2), a severe and progressive cholestatic liver disease, which may lead to development of hepatocellular carcinoma and need for liver transplantation [2]. In contrast, common ABCB11 variants as well as its inhibition have been associated with transient cases of liver injury such as intrahepatic cholestasis of pregnancy, drug-induced liver injury (DILI), sepsis- and parenteral nutrition (PN)-induced cholestasis [3]. Despite ABCB11 variants have been associated with TNC [4, 5], the proof for impaired BSEP function in the pathogenesis of TNC has been missing. In particular, no evidence has been shown for persisting BSEP dysfunction after discontinuation of triggering factors. Hence, here we describe a novel heterozygous ABCB11 variant and provide the histological correlate as a potential molecular mechanism for TNC.
Case presentation
A term male newborn was delivered by emergency cesarean section to a 27-year-old primigravida with gestational diabetes. The mother had followed nutritional recommendations and had taken no medication during pregnancy. The newborn was large-for-gestational-age, had an umbilical arterial pH of 6.92 and an APGAR score of 1 at 1 min. After cardiorespiratory resuscitation, his APGAR scores were 8 and 10 at 5 and 10 min respectively. His blood tests revealed acidosis, hypoglycemia, severe multiorgan involvement and impaired coagulation, requiring whole body cooling, mechanical ventilation, empiric antibiotic treatment, analgesia, sedation, PN, glucose infusions and clotting factor replacement in the first week of life. Subsequently, the newborn’s clinical condition and laboratory findings improved, the whole body cooling and mechanical ventilation were stopped on day 4 and 7 respectively and full oral feeding was reached on day 14. Despite laboratory findings of multiorgan involvement returned to normal, ALT elevation and direct hyperbilirubinemia persisted beyond the age of 3 weeks (Table 1). Biliary atresia, congenital infections, α1-antitrypsin deficiency, thyroid dysfunction, cystic fibrosis and metabolic disorders were excluded. Genetic analysis of the main hepatobiliary transporters discovered a novel heterozygous missense variant in the ABCB11 gene (c.1345G > A, p.Glu449Lys). According to current ACMG guidelines the variant was classified as likely pathogenic [6]. Furthermore, 2 heterozygous variants, both classified as variants of unknown significance were identified in the gene ATP8B1 encoding the biliary aminophospholipid transporter (c.636 T > A, p.Ile212Ile and c.1819 + 49 T > C). Treatment with ursodeoxycholic acid (UDCA) was initiated on day 28. Due to persisting cholestasis, occasional increase of gamma-glutamyl transferase (GGT) and rising levels of liver transaminases (without an obvious reason) a diagnostic liver biopsy was performed at the age of 3.5 months. Liver histology showed giant cell transformation, lobular inflammation, bile retention inside the hepatocytes and bile canaliculi and mild portal fibrosis and ductal proliferation (Fig. 1a, b). These findings resembled histological presentation of PFIC2 [7]. Specifically, identification of bile plugs within the hepatocytes and bile canaliculi suggested dysfunction of bile elimination mechanisms. Interestingly, immunohistochemical staining revealed almost completely abolished canalicular BSEP expression (Fig. 1c). No additional medication was initiated in our patient. His clinical course improved continuously. UDCA therapy was discontinued at the age of 6 months due to normalization of serum bilirubin levels. Serum liver enzymes returned to normal by the age of 14 months. Our patient showed normal physical development and had normal liver ultrasound findings as well as biochemical liver function tests during the follow-up controls performed on the regular basis till the age of 5 years. Since surrogate parameters of hepatocellular injury and cholestasis improved, we believe that BSEP loss was of transient nature and found it unethical to perform a second liver biopsy for the histochemical proof of recovery.

a. and b. Hepatocellular edema, multinucleated hepatocytes (arrows), accumulation of bile pigment inside the bile canaliculi and hepatocytes (black arrowheads) and mild portal infiltration of neutrophil and eosinophil granulocytes (white and grey arrowheads respectively) is presented. Original magnification: 60 x. pv: portal vein, cv: central vein. c. Immunohistochemical stainings for BSEP (BS transporter) and MRP2 (canalicular bilirubin and glutathione transporter) are shown in the control and patient’s liver specimens. As shown, BSEP-positive inclusions retained in the hepatocellular cytoplasm and no transporter was detected on the canalicular membrane. The internal control MPR2 shows preserved canalicular expression. Original magnification 100x. The specificity of immunohistochemical staining was confirmed by negative control (not shown)
Discussion and conclusions
We report on a previously unknown heterozygous ABCB11 variant and provide for the first time the histological proof of BSEP decompensation in TNC after severe neonatal asphyxia. Ischemic liver injury occurs as part of so-called “multi-organ damage” following perinatal asphyxia and becomes manifest biochemically in peaks of LDH and liver enzymes within the first 24–72 h. These elevated parameters usually decrease in the following days and return to normal between 6 and 12 days after the hypoxic event [8]. However, prolonged cholestasis in our patient was inconsistent with a course of asphyxia-related liver injury (Table 1). Moreover, evolution of serum liver tests suggested that in contrast to asphyxia-mediated massive hepatocellular damage immediately after birth, impairment of bile elimination mechanisms were responsible for chronic liver injury (ALT/AP ratio of 1.4 at the age of 3 months vs. ALT/AP ratio of 8 on day 1 post-partum). Genetic analysis determined a new heterozygous variant of ABCB11, a unique BS-specific transporter, which loss cannot be compensated by an alternative export mechanism in humans. Due to limited size of the liver specimen and based on data obtained from the genetic analysis, examination of potential molecular mechanisms causing cholestasis was restricted to studying BSEP as the most likely candidate. Although the identified mutation was found in a heterozygous state and GGT levels did not match with classical BSEP deficiency, histologically BSEP was almost abolished from hepatocyte canalicular membrane in our patient. Since clinical severity of different ABCB11 mutations directly correlates with the amount of mature protein on hepatocellular membrane [2, 3], impaired canalicular BSEP expression is likely to play at least a partial role in the pathogenesis of prolonged cholestasis in our patient. Of note, the mother of our patient carries the same mutation with no cholestatic events in her history, indicating that the heterozygous state of the identified mutation is likely to be sufficient to maintain BS excretion under normal conditions. In addition, occasionally elevated GGT levels in our patient were inconsistent with classical presentation of BSEP deficiency in PFIC2. It can, therefore, be speculated that the complicated neonatal period and multifactorial liver injury played a critical role in transient decompensation of ABCB11 originating from non-mutated gene copy. It still remains unclear which mechanisms are responsible for long-term BSEP inhibition in our patient. BSEP is believed to recycle between the plasma membrane and subapical vesicles and its half-life in the apical membrane ranges from 4 to 6 days [3]. This may explain short-term BSEP decompensation and its rapid recovery after triggering factor has resolved, as reported in cases of hypoxia-, sepsis-, PN-induced cholestasis or DILI. In contrast, mechanisms responsible for persisting BSEP dysfunction long after termination of damaging factors, as observed in our patient, remain so far elusive. Interestingly, prolonged cholestasis has been observed in rare cases of DILI [9]. However, the pathogenesis of DILI is complex and involves -in addition to BSEP inhibition- additional mechanisms [10, 11]. This is supported by the fact that only a very low percent of newborns with drug intake during the complicated neonatal course develop TNC. In this context, indirect inhibition of BSEP by other hepatobiliary transporters through the farnesoid X receptor (FXR)-mediated signaling has to be considered. Despite two heterozygous mutations found in ATP8B1 were of unknown significance, BSEP inhibition through ATP8B1-FXR-mediated mechanism cannot be excluded [12] and ATP8B1 dysfunction should be considered as additional potentially pathogenic mechanism in course of transient cholestasis in our patient. We believe that prolonged BSEP deficiency and resulting intracellular cholestasis were of multifactorial origin in our patient. In this context, persisting hepatocyte inflammatory response triggered by initial massive liver and multiorgan damage and maintained by ongoing interplay of drugs/toxins, mitochondrial inflammatory reaction by accumulating bile acids [13, 14] as well as immaturity of regulatory mechanisms are likely to contribute to BSEP inhibition. However, BSEP inhibition itself, even if secondary to inflammation or toxicity by endogenous intermediates, played important role for the maintenance of prolonged cholestasis in our patient.
In this report, we describe a novel heterozygous ABCB11 variant and provide, for the first time, the histological demonstration of canalicular BSEP deficiency in a patient with complicated neonatal period and TNC. The role of the newly-identified mutation as a potentially predisposing factor for development of cholestasis has to be investigated in future research.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- ALT:
-
alanine aminotransferase,
- AP:
-
alkaline phosphatase,
- AST:
-
aspartate aminotransferase,
- BS:
-
bile salt,
- BSEP:
-
bile salt export pump,
- cv:
-
central vein,
- DILI:
-
drug-induced liver injury,
- FXR:
-
farnesoid X receptor,
- GGT:
-
gamma-glutamyl transferase,
- LDH:
-
lactate dehydrogenase,
- n.m.:
-
not measured,
- PFIC 2:
-
progressive familial intrahepatic cholestasis type 2,
- PN:
-
parenteral nutrition,
- PT:
-
prothrombin time,
- pv:
-
portal vein,
- TNC:
-
transient neonatal cholestasis,
- UDCA:
-
ursodeoxycholic acid
References
Jacquemin E, et al. Transient neonatal cholestasis: origin and outcome. J Pediatr. 1998;133(4):563–7.
Strautnieks SS, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203–14.
Soroka CJ, Boyer JL. Biosynthesis and trafficking of the bile salt export pump, BSEP: therapeutic implications of BSEP mutations. Mol Asp Med. 2014;37:3–14.
Hermeziu B, et al. Heterozygous bile salt export pump deficiency: a possible genetic predisposition to transient neonatal cholestasis. J Pediatr Gastroenterol Nutr. 2006;42(1):114–6.
Liu LY, et al. Association of variants of ABCB11 with transient neonatal cholestasis. Pediatr Int. 2013;55(2):138–44.
Richards S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Evason K, et al. Morphologic findings in progressive familial intrahepatic cholestasis 2 (PFIC2): correlation with genetic and immunohistochemical studies. Am J Surg Pathol. 2011;35(5):687–96.
Muniraman H, et al. Biomarkers of hepatic injury and function in neonatal hypoxic ischemic encephalopathy and with therapeutic hypothermia. Eur J Pediatr. 2017;176(10):1295–303.
Sundaram V, Bjornsson ES. Drug-induced cholestasis. Hepatol Commun. 2017;1(8):726–35.
Aleo MD, et al. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology. 2014;60(3):1015–22.
Chan R, Benet LZ. Measures of BSEP inhibition in vitro are not useful predictors of DILI. Toxicol Sci. 2018;162(2):499–508.
Alvarez L, et al. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004;13(20):2451–60.
Cai SY, et al. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight. 2017;2(5):e90780.
Yang K, et al. Sandwich-cultured hepatocytes as a tool to study drug disposition and drug-induced liver injury. J Pharm Sci. 2016;105(2):443–59.
Acknowledgements
We thank Dr. Bruno Stieger (University of Zurich, Switzerland) for providing us with anti-BSEP antibody.
Funding
There is no funding source to declare.
Author information
Authors and Affiliations
Contributions
AB designed the theory, performed data analysis and wrote the manuscript, LO performed the genetic analysis, CL performed histological examination of biopsy specimen, PF discussed the results, BR, NM and AD contributed to data generation and to the critical revision of the final manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The written informed consent has been obtained from the mother of our patient for publication of the findings (our patient is under 18 years old).
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Baghdasaryan, A., Ofner-Ziegenfuß, L., Lackner, C. et al. Histological demonstration of BSEP/ABCB11 inhibition in transient neonatal cholestasis: a case report. BMC Pediatr 20, 340 (2020). https://doi.org/10.1186/s12887-020-02201-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-020-02201-x