
Abstract
Background
Cytomegalovirus [CMV] is a causative agent of congenital infection worldwide and often leads to neurological deficits and hearing loss in newborns. Infants born with symptomatic congenital Cytomegalovirus infection [cCMV] are at significant high risk for developing adverse long-term outcomes. In this study, we look into the sequence variability of surface glycoprotein B [gB] encoding region in newborns with symptomatic CMV infection for the first time in Eastern region of India.
Methods
576 suspected newborns from seropositive mothers were subjected to the study and ELISA was used to confirm CMV infection. Different genotypes and their subtypes were determined using multiplex nested-PCR. Viral load of different glycoprotein B [gB] genotypes was measured using RT-PCR. Sequencing and phylogenetic analysis was then performed using Bayesian interference.
Results
The overall frequency of cCMV infection was 18.4%, where 16.0% neonates were symptomatic. Among the different gB genotypes, gB1 had the highest frequency [23.5%] and gB4 showed the lowest occurrence [5.8%]. 23.5% of symptomatic neonates had mixed genotypes of gB, probably indicating matrenal reinfection with CMV strains in Indian population. Significant genotypic clades [gB1-gB2-gB3-gB5] were grouped closely based on gene sequences, but the gB4 sequence was in the outlier region of the phylogenetic tree indicating the genetic polymorphism.
Conclusion
This is the first study on cCMV genotyping and its phylogenetic analysis from Eastern Indian neonatal population. The study holds importance in the assessment of cCMV seroprevalence in global perspective. gB protein can be used as a potential therapeutic target against CMV infection.
Similar content being viewed by others


Background
Cytomegalovirus [CMV] has emerged as one of the leading cause of viral congenital infection and is a neglected problem worldwide including India. This is due to the fact that almost all of these congenital infections remain asymptomatic and may not be recognized at birth [1]. The immune status of the patients determines reactivation of latent CMV infection in several clinical cases. Reactivation of latent infection leads to an increased chance of recombination and development of novel CMV variants [2]. Congenital CMV infection is caused due to maternal transmission of new or reactivated latent infection to the fetus, at any gestational stage. Newborns may acquire CMV infection through congenital, intrapartum and antenatal routes of infection. Congenital CMV infection is transmitted transplacentally and may result in symptomatic or asymptomatic infection in neonates. Several epidemiological studies on the incidence of congenital Cytomegalovirus [cCMV] infections have been carried out in many countries of Europe and the United States of America [3]. Some limited number of seroepidemiological studies that has been conducted in the Indian population, show 80–90% prevalence of IgG antibodies in women of childbearing age [4, 5]. Infants with cCMV infection are categorized as symptomatic and asymptomatic based on presence and absence of clinical findings. Around 90% of children with cCMV infection do not show clinical abnormalities and are classified as asymptomatic, whereas only 10% of children showing clinical abnormalities are considered symptomatic [6,7,8,9].
Newborns with symptomatic infections are at severe risk for developing adverse neurodevelopmental sequelae [6]. Symptomatic cCMV infections are associated with intrauterine growth retardation [IUGR] [10], long-term neurological sequelae [i.e., cognitive and motor impairment, hearing loss, visual impairments], microcephaly [11], petechiae, jaundice, hepatosplenomegaly [11, 12], retinitis [4], cerebral abnormalities [13], thrombocytopenia, and fetal/infant death [13, 14]. Prevention and control of cCMV infection stands as an ongoing challenge [12, 15]. Traditional cCMV diagnosis is complicated, as serological evidence of active infection cannot be correlated with the clinical status [9]. The magnitude of the problem in India needs thorough and in-depth investigation. Serological assays for CMV-specific IgM have been previously used in several studies, for detection of congenital infection [1, 5, 16, 17]. Our study has been designed to address the problems of cCMV genetic variations among the neonates of the Eastern Indian region. CMV glycoprotein B [gB] [UL55] is a major envelope glycoprotein, which exists in the viral envelope and acts as proteolytically processed protein dimer on the membrane surface of all CMV-infected cells [18]. Apart from its role in virus entry and fusion, gB is also required for cell-to-cell proliferation. gB gene plays a key role in the “priming” of the transcriptional machinery in the host cell before viral replication begins [19]. Accordingly, gB gene might be a powerful cCMV pathogenicity marker for their potential implication in virulence and cell tropism. The present study is one of the first reports showing the prevalence of cCMV infection in symptomatic neonates of Eastern Indian population. We further examined the distribution of CMV-gB genotypes and it was attempted to associate the clinical and prognostic significance with the circulating genotypes. The study also holds a promising role in utilizing gB as novel marker of infection.
Methods
Study groups and clinical specimens
The study was carried out in the Virology Laboratory [Indian Council of Medical Research Virus Unit, Kolkata, West Bengal, India] between August 2014 and July 2016. In total, 576 live-born infants, born to CMV infected mothers, from different metropolitan hospitals and medical colleges were included in this study. The patients enrolled for the study included suspected newborns within 2 weeks of birth. Infants born to CMV infected mothers having present/active infection [tested positive for IgM] or past infection [tested positive for IgG] was the chosen population for the study. Majority of cases reported in hospitals were mixed population from both urban and rural areas, but mostly from low socio-economic strata.
Sample collection, clinical assessment, screening and serodiagnosis
About 3–5 mL of blood specimens were collected from suspected newborns within 2 weeks after birth. Clinical data were collected from the information sent by the physicians. All serum samples were screened and the presence of CMV-IgM antibody was determined by enzyme-linked immunosorbent assay [ELISA] using the commercially available kit [Equipar SRL, Lombardia, Saronno, Italy] according to the manufacturer’s instructions. Neonates with positive IgM and CMV-DNA in blood within 2 weeks of life with clinical signs and symptoms were considered as symptomatic congenital CMV infection.
Genomic DNA isolation and quantification
CMV DNA was extracted from 200 μl of plasma using the QIAamp DNA Blood MiniKit [QIAgen, Hilden, Germany] following manufacturer’s instructions. Extracted DNA from symptomatic newborns were subjected to PCR for the molecular genotyping through amplification and detection of gB genotypes.
Estimation of viral load using real time PCR
Real-time PCR reactions and quantitative analysis were performed to measure CMV titre in specimens based on the earlier method described by Kubar et al., 2004 [20]. PCR amplifications were performed as individual assays for each sample. The sequences for the PCR primers and probes were designed using the PrimerQuest tool from Integrated DNA Technologies [IDT]. The TaqMan probes were labeled with 6-carboxyfluorescein [FAM] reporter dye at the 5′ end, and with the 6-carboxytetramethyl-rhodamine [TAMRA] quencher dye at the 3′ end as described previously. Amplification, data acquisition and all analyses were carried out using the ABI 7200 SDS [Applied Biosystems, Foster City, CA, United States].
Statistical analysis
All live-born infants were systematically screened during the study period to determine the birth prevalence of symptomatic cCMV infection. The birth prevalence was defined as the number of infected infants divided by the total number of live-born infants. Fisher’s exact test was used to evaluate the statistical impact of different groups, and p values < 0.05 were considered significant at the 95% confidence interval. Statistical analysis was conducted using the Statistical Package for the Social Sciences [SPSS] 16.0 software [SPSS, Inc., Chicago, IL, USA].
Primer designing for conventional CMV PCR
The CMV glycoprotein B [gB] gene sequence was extracted and retrieved from the laboratory strain AD169 [FJS27563] through the NCBI database. All open reading frames [ORF] were reanalyzed and recalculated by selecting the start codon through ORF finder [21, 22]. The most conserved domain was distinguished utilizing the NCBI-conserved domains database [NCBI-CDD] and BLASTp algorithms [23, 24]. CMV-specific internal primers for gB genotype was designed using Primer 3.0 [Table 1]. In the East Indian newborns, sequence variability of gB genotypes [gB1 to gB5] were amplified and analyzed using external primer and internal primers as described by Tarrago´ et al. [2].
Molecular diagnosis of gB genotypes by multiplex nested PCR [M-nPCR]
A standard strain of CMV [ATCC-AD169] was used as the positive control and amplification reactions were performed by the GeneAmp PCR system [Applied Biosystems, USA].
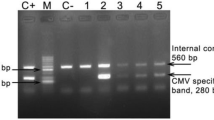
Genotypic distribution of gB gene was measured in 17 symptomatic cCMV infected neonates. For the first-round amplification of glycoprotein B [gB] gene, reaction mixtures consisted 12.5 μL of 2X ready master mixture [Takara, Japan], 5 μl of target DNA [60–65 ng], 10 pmol of each oligonucleotide primer, and molecular grade water [Sigma, USA] to a total volume of 25 μl. The conditions for amplification with external primers were 94 °C for 3 min, followed by 35 cycles of 94 °C for 45 s, 55 °C for 45 s, and 72 °C for 45 s, and the final extension at 72 °C for 5 min. For the second round PCR, 2 μL of each amplicon was added as template to the 12.5 μL of 2X ready master mixture [Takara, Japan], 10 pmol of each internal primer, and molecular grade water [Sigma, USA] to a total volume of 25 μl. The second round PCR program carried out at 94 °C for 3 min, followed by 35 cycles of 94 °C 45 s, 52 °C for 30 s and 72 °C for 45 s with a final extension at 72 °C 5 min. The amplified products were estimated by visual comparison with the standard DNA molecular markers and amplicons were generated after electrophoresis through 2.0% agarose gel, stained with 0.5 mg ml-l ethidium bromide [Sigma Chemicals Co., USA] and visualized on Gel Documentation System [Biorad].
DNA sequencing
Amplified gB gene products from each gB genotype was selected for sequencing from symptomatic newborns. The sequences obtained were visualized as computer traces using Chromas lite v1.62. All sequences were analyzed in both, forward and reverse directions.
Bioinformatics and phylogenetic analysis
The nucleotide sequences of gB gene was checked, edited and analyzed by DnaBaser v3.0. Clustal omega with complete alignment mode was used for the multiple sequence alignment. Our sequences were compared with published sequences from Viper, DDBJ/EMBL/NCBI-GenBank database along with sequences of AD169, Merlin, Toledo, and Towne reference strains. Inter/intra-genetic distribution and their variations were analyzed and compared on local and global perspectives. Phylogenetic analysis was performed through Monte Carlo Markov Chains [MCMC] methods using Bayes’ theorem [25], which incorporated a model of evolution, quantified and addressed the source of uncertainty and was able to incorporate complex models of evolution. Bootstrap calculations were based on 1000 repetitions [26]. Binary matrix was calculated to reconfirm the intra-sequence distribution and polymorphism in different gB-genotypes in symptomatic neonatal samples. Net average distances between groups of taxa was calculated using the formula dA = dXY – [dX + dY]/2; where, dXY is the average distance between groups X and Y, and dX and dY are the mean within-group distances. The average of outgroups is shown with “n/c”. Analyses were conducted using the Poisson correction model [27].
Results
Out of 576 newborns tested within 2 weeks after birth, the prevalence of cCMV infection was 18.4% [106/576] with or without symptoms. Among all infected newborns, 16.0% [17/106] of neonates were symptomatic and rest [n = 89] were asymptomatic [83.9%]. Symptomatic newborns with distinct clinical manifestations were enrolled in this study [Fig. 1]. Gender wise distribution among 17 symptomatic cCMV infections was 29.4% [5/17] in male and 70.5% [12/17] in female. A total of 89 asymptomatic cCMV positive neonates showed a gender-wise distribution of 35.9% males and 64% females. Thus, among a total of 106 CMV affected neonates, 69% were females and 37% were males. A dominance of CMV infection among females was thus established.

Proposed flow diagram showing the strategy for selection of patients study group for the maternal transmission of CMV and diagnostics for identification of congenitally infected neonates with suspected cytomegalovirus
After confirming the CMV infection of newborns, studies on societal history of the families indicated that the cohort was restricted to a low-income group. It was observed that 52.9% [9/17] of mothers were working women. Though there was a higher prevalence of CMV-IgM among working women compared to housewives, the difference did not show significant association between occupation and IgM prevalence. Among clinical manifestations, hepatosplenomegaly was the most common feature [47.0%] followed by neonatal cholestasis/hyperbilirubinemia [41.1%], thrombocytopenia [35.2%], neonatal jaundice [29.4%], and IUGR [23.5%]. 11.7% of neonates were affected with bronchopneumonia [n = 3] and septicemia [n = 3] [Table 2]. Congenital cataract and hearing impairment was seen in 1 infant each. CT scan of the brain was performed in 3 infants as suggested by physicians and single infant showed bilateral sulcal calcification along with cholestatic jaundice and this particular finding was reported previously by our group [28].
CMV load was determined in 17 gB genotyped symptomatic cCMV infected samples [Table 3]. The overall median load of gB was 3.9 × 104 log10/ml. Similarly, gB genotyped viral load was 2.1 × 103 log10/ml for gB1, 2.4 × 104 log10/ml for gB2, 3.8 × 103 log10/ml for gB3, 3.1 × 104 log10/ml for gB4 and 2.2 × 103 log10/ml for gB5. Viral load of mixed genotypes were 3.9 × 103 log10/ml, 3.9 × 104 log10/ml, 4.8 × 104 log10/ml for gB1, gB2; gB1, gB3; and gB2, gB5 respectively [Fig. 2a]. Genotypic prevalence of gB protein showed gB1 had highest frequency, followed by gB2 and gB3 while gB4 showed negligible occurrence [Fig. 2b]. Genotyping based on the gB region was determined by PCR from CMV-positive cultures where gB external primers was 100% positive in all cases [17/17]. In addition, the M-nPCR assay for gB genotyping was able to detect all samples containing single genotypes and as well as a mixture of genotypes. Table 3 summarizes the frequency of gB genotypes according to specimens along with clinical manifestations. The single genotype was distributed in 13 [76.47%] samples. gB1 was the most frequently occurring genotype and observed in 4 specimens [23.52%]. While 17.64% of gB2 and gB3 genotype was observed [n = 3] in each genotype, a single sample corresponded to gB4 [5.8%] and two for gB5 [11.76%]. However, a mixed-genotype infection was also detected in 4 [23.52%] cases. Among these mixed genotype infections, all were dual gB genotype infections and included 11.76% [2/17] gB1 and gB2, 5.88% [1/17] of gB1-gB3 and gB2-gB5 genotypes.

[a] Distribution of CMV-DNA load in congenitally infected symptomatic neonates. CMV DNA load of different gB genotypes and their subtypes was quantify using real time-PCR. Estimation of gB genotypes were corelated in five different subtypes. [b] Schematic representation for the genotypic distribution of CMV-gB pure and mixed subtypes in symptomatic newborns infected with CMV
Submitted sequences of gB gene were compared to determine inter/intra genomic variations. GenBank accession numbers of the representative submitted sequences for 17 gB gene region from this study are KY436004 to KY436020. Sequences from the variable region of gB gene was compared to the published sequences of all gB genotypes. BLASTn analysis of our nucleotide sequences revealed about 98–100% similarity to prototype strains. Corresponding peptide sequences were translated using EMBOSS-Transeq tool [http://www.ebi.ac.uk/Tools/st/emboss_transeq/] and analyzed with the reference strains and presented in Fig. 3a and b.

Amino acid sequence alignment of distinct sequences identified in the study. [a] Sequence alignment of gB4 genotype with the prototype strain, AD169. [b] Universal alignment of all the glycoprotein [gB] genotypes [gB1–5] with prototype strain, AD169
Similarity index was calculated through multiple sequence alignment [MSA]. Finally, the gB gene clusters and comparative sequences were analyzed based on the theory of Bayesian Probabilistic Approach 23, 24]. A total of 10,000 generations was taken for phylogenetic tree, which provided the bootstrap values in each branch after the analysis using Bayesian posterior probabilities, and was proportional to the number of substitutions per site. Different gene clusters were found with considerable distance, and each cluster was grouped within the same genotype except gB4 [Fig. 4a and b]. The evolutionary history was obtained by applying Neighbour-Join and BioNJ algorithms to a matrix of pairwise distances. The tree was drawn to scale, with branch lengths measured in the number of substitutions per site with all sequences. All nucleotide sequences were translated and compared within intra genotypic variations. Along with previously published sequences, five genotypic clades were identified in phylogenetic analysis. The phylogenetic tree indicated that genotype gB4 was distant, while genotypes gB1 with gB2 and gB3 with gB5 were more closely related [Fig. 4a]. Sequence comparison of amino acids in different types of symptoms-associated genotypes and predicted sequence alignment revealed that each of genotype was mostly homologous to each other [Fig. 4b]. It was observed that gB4 sequence had maximal significant variations among all the gB genotypes. The genotypic distribution was significantly variable from different geographical regions [29]. In Asia, Japanese genotypic distribution was found similar to the European genotypes [30, 31], while Chinese population showed a different pattern [32].

Phylogenetic tree based on the Neighbour-joining [N-J] method for the analysis of CMV gB gene sequences. [a] The intra genetic distribution along with reference strains; and the phylogenetic position of CMV-gB sequences isolated from eastern India and [b] Evolutionary relationships of taxa, where the optimal tree with the sum of branch length = 3.59972651 is shown. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test [1000 replicates] are shown next to the branches. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. The analysis involved 65 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All positions containing gaps and missing data were eliminated. [Ο represents CMV-AD169 reference strain and Δ represents isolates belonging to cCMV patients]
We further tried to compare the genotypic distribution of gB with the different clinical manifestations observed in the patients. Hepatosplenomegaly, neonatal cholestasis and thrombocytopenia was observed in 4 (23.52%) symptomatic patients with genotype gB1. Genotype gB2 was associated with IUGR and microcephaly in 3 (17.64%) infected infants. gB3 and gB5 was found in 3 (17.64%) and 2 (11.76%) patients respectively, who were suffering from septicemia and bronchopneumonia. Genotype gB4 was observed in 1 (5.8%) patient and was possibly related to various neurological disorders [e.g., Congenital cataract, hearing impairment] along with microcephaly and hepatosplenomegaly [Table 3]. This is a preliminary study conducted in a small cohort and needs to be conducted in a larger population to confirm the association of gB genotypes with specific clinical manifestations from Eastern India. Calculated binary matrix for each group was computed for all valid pairwise comparisons and results are displayed in the Table 4, where most polymorphic genotype [gB4- KY436019] belonged to the outgroup within cluster IV and matrix showing incalculable within group and shown as “n/c”.
Discussion
cCMV is a leading cause of public health problem throughout the world including India. Asymptomatic infants affected with CMV infection have no apparent clinical symptoms and 10–15% of these children develop long-term sequelae [33,34,35]. The prevalence of congenital CMV infection may vary on the basis of the differences in a geographical region, depending on the racial, ethnic, socioeconomic background and diagnostic methods used to detect infection [36]. Due to a high seroprevalence of CMV in developing countries, diagnosis of cCMV infection in symptomatic patients is recommended to reduce infant morbidity, mortality and sequels [37]. Very limited information is available about the incidence and the history of this infection in India [4, 38]. There are no previous reports on molecular data for symptomatic cCMV infection from Eastern Indian region. This is the first genotyping study of cCMV-gB transmission among symptomatic neonates in Eastern India region, to the best of our knowledge. Numerous studies reported around 10–15% of neonates show relevant clinical signs with congenital infection during birth [39]. Our study showed that congenital CMV infection had a high prevalence in Eastern India, which was in corroboration with other parts of India and abroad. Since the symptomatic newborns are at a much higher risk for developing clinical complications, we chose to study the genotypic variation in this population. The asymptomatic newborns are at a lower risk for adverse neurodevelopmental sequelae and thus they may be considered for a follow-up study, to ensure a better management of asymptomatic disease burden. The present study shows that the prevalence of symptomatic cCMV infection was 16.03% from total CMV positive newborns. Kenneson and Cannon reported the overall birth prevalence of congenital CMV infection was 11.0% and varied considerably among different study populations [40]. Several studies also reported that about 13.5% of CMV infected newborns are born with symptoms [33, 41]. Another study has reported that 19.4% of babies were congenitally infected through CMV infection with various birth defects in India [29]. In this study, gender wise prevalence was measured in symptomatic as well as asymptomatic newborns. 29.4% males and 70.5% females were infected with CMV among symptomatic newborns. On the other hand, 35.9% males and 64.0% females were found asymptomatic. In both cases [symptomatic and asymptomatic], distribution frequency was higher in female patients as compared to male. Most frequently observed clinical symptoms were hepatosplenomegaly [47%] and neonatal cholestasis [41.1%], which was mostly associated with the gB1 genotype. These observations are in concordance with other previous reports [39, 40] [42, 43]. Shukla et al., [2015] found a significant pre-dominance of females (59.72%) among CMV positive patients [44]. Similar results were stated by Colugnati et al., [2007], where CMV IgG titre was higher in females [45]. A report by Firth et al., 2016 and Lachmann et al., 2018 also explained the higher incidence of CMV in female patients [46, 47]. Genetic polymorphisms of envelope glycoproteins among circulating CMV strains are generally considered as probable virulence indicators [48], and may be accountable for differential CMV tropism to precise cell types and differential capacity to distribute and interfere with normal tissue growth [30, 39]. Previous reports show that four gB genotypes of CMV are observed in congenital cytomegaly [48, 49]. Several studies reported that cCMV infections among Costa Rican, Indian and Chinese infants were caused by gB1, gB2, and gB3 genotypes [29, 42]. In a Polish study, cCMV infection occurrence of gB1, gB2, gB3, and gB4 genotypes was found to be about 43.7, 31.25, 25.00, and 12.5%, respectively [50]. A previous study showed high prevalence of gB1 and gB2 genotypes [50.0%], reduced prevalence of gB3 genotype [8.3%] and negligible incidence of gB4 genotype in affected subjects [49]. Our studies revealed the highest incidence of gB1 [23.5%], moderate incidence of gB2 and gB3 [17.6%], partial incidence of gB5 [11.7%] and negligible occurrence of gB4 [5.8%] among symptomatic neonates of eastern India. Interestingly, this was in concordance with genotypic distribution of cCMV infection from different geographical regions including Costa Rica, rest of India, China and South Hungary and also had gB1 as the most prevalent circulating genotype in these regions [4, 39, 42, 51]. Differential distribution of envelope glycoprotein genotypes circulating in discrete geographical regions among congenitally infected patients have been reported previously [2, 3]. Interestingly our studies revealed that in eastern India, gB1 genotype for monoinfection and gB1-gB2 for mixed infection is maximally prevalent in cCMV infection. Similar reports were observed in other studies [3]. Our study reports genotypic distribution of cCMV infection in Eastern Indian region for the first time. We report that 23.5% of symptomatic neonates had mixed genotypes of gB. This is an interesting finding and previous reports show occurrence of mixed genotypes of gB in geographical regions other than India, which we have discussed vividly. A probable reason for this finding suggests that maternal reinfection with CMV strains could be frequent in Indian populations. It has been observed previously, that presence of multiple gB genotypes could be a critical factor associated with severe clinical manifestations compared to the presence of a single gB genotype, in immunocompromised patients [52].
Further, it was observed that the symptomatic neonates with mixed genotype infection had detectable appreciable viral load. Previous studies have shown that infections with mixed genotypes increases the chances of progression to CMV diseases [52, 53].
Among the different circulating glycoprotein genotypes in symptomatic newborns of eastern India, gB1 was maximally prevalent followed by gB2 and gB3, while gB4 showed negligible occurrence. Similar studies reported that gB1 was the more frequent genotype in infants infected congenitally in Hungary [39], Italy [51, 54] Japan] [30, 31], and the United States [50]. gB2 was found to be a major genotype transmitted congenitally in Australia [55]. Picone et al. also found a similar distribution of genotypes gB1, gB2, and gB3 in CMV strains recovered from amniotic fluid samples in France [56].
Intra-host viral diversity and genomic variation have recently been characterized by next-generation sequencing, showing similar genomic variability in different RNA viruses [48, 49] [57, 58]. Mixed populations of the glycoprotein genotype have been previously documented in congenitally infected neonates and in pregnant women [59]. Our study describes the primary circulating genotypes in the affected neonates, and these genotypes probably play vital roles in disease severity in this geographical region.
As the genotypic distribution of CMV varies among different geographical regions [60], we compared the symptomatic cCMV-gB genotypes with those reported previously. Present study was formulated to address the polymorphisms of CMV genes encoding different gB genotypes in symptomatic cCMV infections. Results from this study was further analyzed and confirmed through DNA sequencing and phylogenetic analysis using Bayesian interference. According to the algorithm for sequence comparison, gB variable region was compared to the other available repository sequences in the database. Phylogenetic analysis from our study suggested that gB1 and gB2 may have common evolutionary origin in Eastern Indian region as both are clustered together in the same clade. However, gB3 and gB5 gene cluster was found in the different clade with considerable distance. The four clusters [gB1, gB2, gB3 and gB5] were grouped within the same genotype except gB4. In our study, we observed that gB4 positive neonates were related to specific neurological disorders like congenital cataract and hearing impairment. These particular findings are different from the study which was conducted in the other part of India, where they have found that same symptoms were related to gB2 genotype [27]. Comparison of the sequences on global perspective portrayed a distinct pattern of gB genotypes, where eastern Indian sequence was clustered within the same genotype along with other gB genes of various province. Interestingly, this revealed that CMV gB gene is composed of highly conserved regions.
This is the first report on genotypic variations in cCMV infection from eastern India region. gB4 genotypic polymorphisms have been associated with variable clinical outcomes. Our study focuses on the initial correlation of the genotype variability with the different diseases in symptomatic neonates from eastern India. CMV is able to establish latency and evade immune surveillance. This presents particular challenges in the development of effective vaccination as CMV genome displays great genetic heterogeneity. The purified gB protein stands as an important candidate for vaccine development against CMV infection. Neutralizing antibodies against CMV infection was produced when the purified protein was injected in combination with MF-59 oil-in-water adjuvant in humans for a fixed regimen of six months [34]. Thus, there is a constant need for upgrading the information on molecular epidemiology of CMV in different population and geographical regions, which would help in developing effective universal vaccines for the prophylactic treatment of CMV in humans.
Conclusion
We believe to the best of our knowledge that this is the first molecular evaluation on cCMV-gB genotyping and their phylogenetic position in east Indian symptomatic neonatal population. This is important for assesement of the cCMV seroprevalence in the global perspective. This study also highlighted the efficacy of PCR-based techniques in following possible transmission of CMV within communities. However, it is worth mentioning that we had limited sample size for the seroprevalence screening. Taken together, the present study is extremely useful in developing gB protein as a prospective therapeutic target for CMV. We strategize to carry out a follow up study for the asymptomatic cCMV infected newborns to have a bird’s eye view of the disease in the global perspective. Similar disease specific genes can be identified for developing curative approaches for other viral infections.
Availability of data and materials
Complete raw data is available on request. Kindly contact the corresponding authors for raw data.
Abbreviations
- ATCC-AD169:
-
American Type Culture Collection-AD169 cytomegalovirus strain
- BLASTn:
-
Basic Local Alignment Search Tool for nucleotide databases
- cCMV:
-
Congenital Cytomegalovirus
- CDD:
-
Conserved domains database
- CMV:
-
Cytomegalovirus
- DDBJ:
-
DNA Data Bank of Japan
- EMBL:
-
European Bioinformatics Institute
- gB:
-
Glycoprotein B
- IUGR:
-
Intrauterine growth retardation
- MCMC:
-
Markov Chains
- NCBI:
-
National Center for Biotechnology Information
- RT-PCR:
-
Real time PCR
- UL55:
-
unique long gene 55
References
Ebrahim MG, Ali AS, Mustafa MO, Musa DF, El Hussein ARM, Elkhidir IM, Enan KA. Molecular detection of human Cytomegalovirus [HCMV] among infants with congenital anomalies in Khartoum state. Sudan The Open Virol J. 2015;9:38–41.
Tarrago D, Quereda C, Tenorio A. Different Cytomegalovirus Glycoprotein B Genotype Distribution in Serum and Cerebrospinal Fluid Specimens Determined by a Novel Multiplex Nested PCR. J Clin Microbiol. 2003;41(7):2872–7.
Murph JR, Souza IE, Dawson JD, Benson P, Petheram SJ, Pfab D, Gregg A, O'Neill ME, Zimmerman B, Bale JF. Epidemiology of congenital cytomegalovirus infection: maternal risk factors and molecular analysis of cytomegalovirus strains. Am J Epidemiol. 1998;147(10):940–7.
Chakravarty A, Kashyap B, Rathi K. The seroepidemiological study on cytomegalovirus in women of child-bearing age with special reference to pregnancy and maternal-fetal transmission. Indian J Pathol Microbiol. 2005;48:518–21.
Gandhoke I, Aggarwal R, Lal S, Khare S. Congenital CMV infection in symptomatic infants in Delhi and surrounding areas. Indian J Pediat. 2006;73:1095–7.
Boppana SB, Ross SA, Fowler KB. Congenital Cytomegalovirus infection: clinical outcome. Clin Infect Dis. 2013;57:S178–81.
Britt WJ. Cytomegalovirus. In: Remington JS, Klein JO, Wilson CB, Nizet V, Maldonado Y, editors. Infectious diseases of the fetus and newborn infant. Philadelphia: Elsevier Saunders; 2011. p. 706–55.
Townsend CL, Forsgren M, Ahlfors K, Ivarsson SA, Tookey PA, Peckham CS. Long-term outcomes of congenital cytomegalovirus infection in Sweden and the United Kingdom. Clin Infect Dis. 2013;56:1232–9.
Bonalumi S, Trapanese A, Santamaria A, D’Emidio L, Mobili L. Cytomegalovirus infection in pregnancy: review of the literature. J Prenat Med. 2011;5:1–8.
Angélica LD, Alicia A, Fabián M, Fabián P. Human Cytomegalovirus: detection of congenital and perinatal infection in Argentina. BMC Pediat. 2004;4:11.
Fowler KB, McCollister FP, Dahle AJ, Boppana S, Britt WJ, Pass RF. Progressive and fluctuating sensorineural hearing loss in children with asymptomatic congenital cytomegalovirus infection. J Pediatrics. 1997;4:624–30.
Boppana SB, Rivera LB, Fowler KB, Mach M, Britt WJ. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N Engl J Med. 2001;344(18):1366–71.
Friedman S, Ford-Jones EL. Congenital cytomegalovirus infection: an update. Pediatr Child Health. 1999;4:35–8.
De Vries JJ, Korver AM, Verkerk PH, Rusman L, Claas EC, Loeber JG, Kroes ACM, Vossen ACTM. Congenital cytomegalovirus infection in the Netherlands: birth prevalence and risk factors. J Med Virol. 2011;83:1777–82.
Barkai G, Ari-Even Roth D, Barzilai A, Tepperberg-Oikawa M, Mendelson E, Hildesheimer M, Kuint J. Universal neonatal cytomegalovirus screening using saliva-report of clinical experience. J Clin Virol. 2014;60:361–6.
van Zuylen WJ, Hamilton ST, Naing Z, Hall B, Shand A, Rawlinson WD, Revello MG, Gerna G. Congenital cytomegalovirus infection: clinical presentation, epidemiology, diagnosis and prevention. Obstet Med. 2014;7:140–6.
Revello MG, Gerna G. Diagnosis and management of human cytomegalovirus infection in the mother, fetus, and newborn infant. Clin Microbiol Rev. 2002;15:680–715.
Wille PT, Wisner TW, Ryckman B, Johnson DC. Human cytomegalovirus [HCMV] glycoprotein gB promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio. 2013;4(3):e00332–13.
Isaacson MK, Compton T. Human Cytomegalovirus glycoprotein B is required for virus entry and cell-to-cell spread but not for Virion attachment, assembly, or egress. J Virol. 2009;83(8):3891–903.
Kubar A, Saygun I, Yapar M, Özdemir A, Slots J. Real-time PCR quantification of cytomegalovirus in aggressive periodontitis lesions using TaqMan technology. J Periodontal Res. 2004;39:81–6.
Sarkar A, Chatterjee A, Ansari S, Chakraborty N. Characterization of molecular mimicry between UL18 glycoprotein of human Cytomegalovirus [HCMV] and class-I MHC molecule through pattern-based analysis: an In-silico approach. J Health Med Informat. 2016;7:230.
Zanet J, Benrabah E, Li T, Pelissier-Monier A, Chanut-Delalande H, Rosin B, Bellen HJ, Payre F, Plaza S. Pri sORF peptides induce selective proteasome-mediated protein processing. Science. 2015;349(6258):1356–8.
Geer LY, Domrachev M, Lipman DJ, Bryant SH. CDART: protein homology by domain architecture. Genome Res. 2002;12:1619–23.
Marchler-Bauer A, Derbyshire MK, Gonzales NR, Lu S, Chitsaz F, Geer LY, Geer RC, He J, Gwadz M, Hurwitz DI, Lanczycki CJ, Lu F, Marchler GH, Song JS, Thanki N, Wang Z, Yamashita RA, Zhang D, Zheng C, Bryant SH. CDD: NCBI’s conserved domain database. Nucleic Acids Res. 2015;43:D222–6.
Larget B, Simon DL. Markov chain Monte Carlo algorithms for the Bayesian analysis of phylogenetic trees. Mol Biol Evol. 1999;16:750–9.
Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5.
Zuckerkandl E, Pauling L. Evolutionary divergence and convergence in proteins. Edited in evolving genes and proteins by V. Bryson and H.J. Vogel, 1965;97-166. Academic press, N Y.
Chatterjee A, Mishra L, Sarkar A, Chatterjee RP, Ansari S, Bhattacharya M, Chakraborty N. Diagnostic dilemma in reckoning cytomegaloviral infection is prompting increased mortality among abandoned newborns of India. Int J Med Res Healt Sci. 2016;5:43–7.
Gandhoke I, Akhtar Hussain S, Pasha ST, Chauhan LS, Khare S. Glycoprotein B genotyping in congenital/perinatal cytomegalovirus infection in symptomatic infants. Ind Pediat. 2013;50(15):663–7.
Murayama T, Takegoshi M, Tanuma J, Eizuru Y. Analysis of human cytomegalovirus UL144 variability in lowpassage clinical isolates in Japan. Intervirol. 2005;48(2–3):201–6.
Yan H, Koyano S, Inami Y, Yamamoto Y, Suzutani T, Mizuguchi M, Ushijima H, Kurane I, Inoue N. Genetic variations in the gB, UL144 and UL149 genes of human cytomegalovirus strains collected from congenitally and postnatally infected Japanese children. Arch Virol. 2008;153(4):667–74.
Mao ZQ, Huang Y, Sun M, Ruan Q, Qi Y, He R, Huang YJ, Ma YP, Ji YH, Sun ZR, Gao H. Genetic polymorphism of UL144 open reading frame of human cytomegalovirus DNA detected in colon samples from infants with Hirschsprung’s disease. World J Gastroenterol. 2007;13(32):4350–4.
Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17:355–63.
Plotkin SA, Boppana SB. Vaccination against the human cytomegalovirus. Vaccine. 2018; [In Press]. https://doi.org/10.1016/j.vaccine.2018.02.089.
Jückstock J, Rothenburger M, Friese K, Traunmüller F. Passive immunization against congenital Cytomegalovirus infection: current state of knowledge. Pharmacol. 2015;95:209–17.
Vaudry W, Rosychuk RJ, Lee BE, Cheung PY, Pang XL, Preiksaitis JK. Congenital cytomegalovirus infection in high-risk Canadian infants: report of a pilot screening study. Can J Infect Dis Med Microbiol. 2010;2(1):e12–9.
Marin LJ, de Carvalho Cardoso ES, Bispo Sousa ES, de Carvalho LD, Marques Filho MF, Raiol MR, Gadelha SR. Prevalence and clinical aspects of CMV congenital infection in a low-income population. Virol J. 2016;13:148.
Mussi-Pinhata MM, Yamamoto AY, Moura Brito RM, de Lima Isaac M, de Carvalho e Oliveira PF, Boppana S, Britt WJ. Birth prevalence and natural history of congenital Cytomegalovirus infection in a highly Seroimmune population. Clin Infect Dis. 2009;49(4):522–8.
Jin H, Wang X, Li S. Human cytomegalovirus glycoprotein B genotype correlates with different symptoms of infected infants. Intervirol. 2007;50(3):219–23.
Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus [CMV] infection. Rev Med Virol. 2007;17(4):253–76.
Yu ZS, Zou CC, Zheng JY, Zhao ZY. Cytomegalovirus gB genotypes and clinical features in Chinese infants with congenital infections. Intervirol. 2006;49(5):281–5.
Lukácsi A, Tarodi B, Endreffy E, Bábinszki A, Pál A, Pusztai R. Human cytomegalovirus gB genotype 1 is dominant in congenital infections in South Hungary. J Med Virol. 2001;65(3):537–42.
Paradowska E, Studzińska M, Suski P, Kasztelewicz B, Wiśniewska-Ligier M, Zawilińska B, Gaj Z, Nowakowska D. Human cytomegalovirus UL55, UL144, and US28 genotype distribution in infants infected congenitally or postnatally. J Med Virol. 2015;87(10):1737–48.
Shukla S, Singh P, Tidke P, Bhatia V, Dutt S. Prevalence of HCMV in Indian Scenario. Int J Pharm Res & Rev. 2015;4(1):1–4.
Colugnati Fernando AB, Staras Stephanie AS, Dollard Sheila C, Cannon MJ. Incidence of cytomegalovirus infection among the general population and pregnant women in the United States. BMC Infect Dis. 2007;7:71.
Firth C, Harrison R, Ritchie S, Wardlaw J, Ferro CJ, Starr JM, Deary IJ, Moss P. Cytomegalovirus infection is associated with an increase in systolic blood pressure in older individuals. QJM: An Int J Med. 2016;109(9):595–600.
Lachmann R, Loenenbach A, Waterboer T, Brenner N, Pawlita M, Michel A, Thamm M, Poethko-Müller C, Wichmann O, Wiese-Posselt M. Cytomegalovirus (CMV) seroprevalence in the adult population of Germany. PLoS One. 2018;13(7):e0200267.
Arcangeletti MC, Simone RV, Rodighiero I, De Conto F, Medici MC, Martorana D, Chezzi C, Calderaro A. Combined genetic variants of human cytomegalovirus envelope glycoproteins as congenital infection markers. Virol J. 2015;12:202.
Zhang MG, Wang HB, Wang YZ, Pan Q. Human cytomegalovirus glycoprotein B genotypes in congenitally infected neonates. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi. 2011;25(4):262–4.
Rycel M, Wujcicka W, Zawilińska B, Paradowska E, Suski P, Gaj Z, Wilczyński J, Leśnikowski Z, Nowakowska D. Mixed infections with distinct cytomegalovirus glycoprotein B genotypes in polish pregnant women, fetuses, and newborns. Eur J Clin Microbiol Infect Dis. 2015;34(3):585–91.
Barbi M, Binda S, Caroppo S, Primache V, Dido P, Guidotti P, Corbetta C, Melotti D. CMV gB genotypes and outcome of vertical transmission: study on dried blood spots of congenitally infected babies. J Clin Virol. 2001;21(1):75–9.
Coaquette A, Bourgeois A, Dirand C, Varin A, Chen W, Herbein G. Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin Infect Dis. 2004;39(2):155–61.
Puchhammer-Stöckl E, Görzer I, Zoufaly A, Jaksch P, Bauer CC, Klepetko W, Popow-Kraupp T. Emergence of multiple cytomegalovirus strains in blood and lung of lung transplant recipients. Transplant. 2006;81(2):187–94.
Arista S, De GS, Giammanco GM, Di CP, Iannitto E. Human cytomegalovirus glycoprotein B genotypes in immunocompetent, immunocompromised, and congenitally infected Italian populations. Arch Virol. 2003;148(3):547–54.
Trincado DE, Scott GM, White PA, Hunt C, Rasmussen L, Rawlinson WD. Human cytomegalovirus strains associated with congenital and perinatal infections. J Med Virol. 2000;61(14):481–7.
Picone O, Costa JM, Leruez-Ville M, Ernault P, Olivi M, Ville Y. Cytomegalovirus [CMV] glycoprotein B genotype and CMV DNA load into the amniotic fluid of infected fetus. Prenat Diagn. 2004;24(12):1001–6.
Sijmoms S, Van Ranst M, Maes P. Genomic and functional characteristics of human cytomegalovirus revealed by next-generation sequencing. Viruses. 2014;6(3):1049–72.
Renzette N, Bhattacharjee B, Jensen JD, Gibson L, Kowalik TF. Extensive genome-wide variability of human cytomegalovirus in congenitally infected infants. PLoS Pathog. 2011;7(5):e1001344.
Brañas P, Blázquez-Gamero D, Galindo A, Prieto C, Olabarrieta I, Cuadrado I, Folgueira L. Cytomegalovirus genotype distribution among congenitally and postnatally infected patients: association of particular glycoprotein [g] B and gN types with symptomatic disease. Open Forum Infect Dis. 2015;2(4):ofv151.
Pignatelli S, Lazzarotto T, Gatto MR, Dal Monte P, Landini MP, Faldella G, Lanari M. Cytomegalovirus gN genotypes distribution among congenitally infected newborns and their relationship with symptoms at birth and sequelae. Clin Infect Dis. 2010;51(1):33–41.
Acknowledgements
We thank Virus Unit [NICED-ICMR], Kolkata, West Bengal, India for providing laboratory facilities, high-speed internet and computational laboratory facilities. We are also thankful to the different metropolis hospitals and medical colleges for providing us samples and patient details.
Funding
Dr. Agniswar Sarkar acknowledges the financial support for Postdoctoral research from the University Grants Commission [UGC], Govt. of India [PDFSS-2013-14-SC-WES-6553]. The contingency grant was used for the purchase of chemicals, used in the different experiments. No additional grants were available to support the research and article processing charges.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: AS, NC. Performed the experiments: AS, RPC, SA. Analyzed the data: AS. Contributed reagents/materials/analysis tools: AS, LM, BB, SKG, MB, NC. Wrote the paper: AS, NC, DD. Samples supplied and detailed information of patients provided by LM, BB, SKG, MB. All the authors have read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study and methodologies were approved by 14th Scientific Advisory Committee [SAC] of Indian Council of Medical Research Virus Unit, Kolkata and certified by Institutional Ethics Committees [IEC] of Dr. B.C. Roy Post Graduate Institute of Paediatric Sciences [Memo no: BCH/ME/PR/2657A]. Written informed consents were taken after explaining all associated positive and negative aspects regarding the study to the guardian of each participating patients, who were under the age of 16, in a language that they could comprehend easily [Bengali, A local language; Hindi, National language; and English, International language].
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Sarkar, A., Das, D., Ansari, S. et al. Genotypes of glycoprotein B gene among the Indian symptomatic neonates with congenital CMV infection. BMC Pediatr 19, 291 (2019). https://doi.org/10.1186/s12887-019-1666-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12887-019-1666-5